La Tétralogie de Fallot – La Maladie Bleue
Amine LABOUDI
Les communications interventriculaires ont toujours constitué la forme la plus commune des cardiopathies congénitales. Cependant, l’une d’elles attire beaucoup plus d’attention que les autres, c’est la tétralogie de Fallot. Une pathologie diversifiée et caractérisée aussi bien par des mécanismes physiopathologiques simples que complexes. Étant la cardiopathie cyanogène la plus fréquente au monde, nous y attachons une importance qui ne saurait être chiffrée. Depuis que la première réparation chirurgicale de la tétralogie de Fallot a été effectuée, il y a une soixante-dizaine d’années, une révolution est mise en feu. Sans relâche, on ne cesse d’attiser ses flammes ; la littérature scientifique en rapport avec cette maladie s’en trouve en croissance perpétuelle.
C’est pour cela que, tout au long du présent article, nous tâcherons, ensemble, de fusiller cette pathologie par des oeillades consciencieuses. Nous aborderons les toutes dernières nouveautés de son aspect génétique, qui lui confère son trait de « maladie congénitale ». Nous expliquerons par la suite les principaux mécanismes physiopathologiques bétonnant ses quatre piliers principaux, desquels elle tient son fameux nom de « Tétralogie ». Puis nous creuserons plus profondément dans ses conséquences hématologiques et hémodynamiques, pour terminer avec une brève revue des moyens diagnostiques et thérapeutiques utilisés pour lui faire face.
Introduction
La tétralogie de Fallot est une maladie qui pose des challenges scientifiques continus sur les deux plans, technique et intellectuel, plus particulièrement vis-à-vis de sa variabilité étendue sur plusieurs aspects. Les docteurs Lev et Eckner ont fait allusion à cela en disant qu’il n’existe pas deux tétralogies de Fallot qui se ressemblent, même si leurs caractéristiques anatomiques permettent de les classer comme une entité phénotypique unique.7, 8
La maladie représente une référence dans la cardiologie pédiatrique étant donné que c’est la première lésion cyanotique qui a été décrite.1 Sa première évocation remonte à 1671 par Niel Stenson, il s’en est suivi, en 1784, l’illustration de la première description anatomique de la maladie par William Hunter, qui l’a exposée ainsi : «… l’orifice de transit du ventricule droit vers l’artère pulmonaire, dont le diamètre devrait être suffisant pour laisser passer un doigt à travers, n’était même pas aussi large qu’une plume d’oie, et il y avait un trou dans la partition des deux ventricules, assez grand qu’un pouce peut entrer dedans et passer d’un côté à l’autre. La majorité du sang était évacuée ainsi du ventricule droit au ventricule gauche, et de là vers l’aorte, ce qui fait que le sang perdait tout avantage qu’il devrait avoir de la respiration…»9, 10 Fallot a contribué par la suite, en 1888, avec son explication de la physiologie résultante de la combinaison d’une communication interventriculaire (CIV) avec une sténose pulmonaire et sous-pulmonaire, tout cela dans sa description de « L’anatomie pathologique de la maladie bleue », faisant référence, avec cette dénomination, à la cyanose qui caractérise la pathologie qui fut nommée plus tard, la tétralogie de Fallot.1 Cependant, le terme « Tétralogie » n’a été attribué à cette maladie qu’avec l’arrivée de Maude Abbot en 1924. Ce dernier a concrétisé les quatre piliers sur lesquels se fonde la tétralogie : (1) La communication interventriculaire ; (2) Le rétrécissement pulmonaire ; (3) La dextroposition de l’aorte ; (4) L’hypertrophie du Ventricule Droit.1
De nos jours, la tétralogie de Fallot compte une incidence de 4 à 5 cas sur 100.000 nouveau-nés. Elle représente 7 à 10% des communications interventriculaires congénitales, ce qui en fait la plus fréquente dans sa catégorie.6
La compréhension des mécanismes menant à l’apparition des quatre piliers sus-cités a permis d’améliorer la prise en charge thérapeutique des sujets qui en sont atteints, mais en contrepartie, les risques de complications à long terme, chez les sujets ayant bénéficié du traitement, ne cessent d’augmenter vu qu’on ne peut pas, jusqu’à maintenant, prédire exactement ce qu’ils pourront expérimenter avec une espérance de vie accrue. Une conclusion définitive des études par rapport à ce sujet n’apparaît toujours pas dans l’horizon.11
La T4F, On Plonge dans la Génétique !
L’aspect génétique de la tétralogie de Fallot est d’une importance capitale. Cependant, la littérature scientifique reste encore limitée à ce propos étant donné la complexité du développement embryonnaire du cœur. Le nombre indéfini de facteurs de transcription et de signal intégrés dans le processus de la cardiogenèse rendent la tâche difficile à assumer, surtout que des gènes additionnels sont découverts continuellement.3 La tétralogie de Fallot survient autant chez les hommes que les femmes.5 Il est généralement admis que l’apparition de la maladie est sporadique, et non pas familiale.5 Toutefois, bien que la descendance des sujets atteints de la TF soit minoritaire, et donc difficile à étudier, on a remarqué une incidence augmentée des cardiopathies congénitales chez ces personnes par rapport à la population générale.12, 13 Même si la base de données sur la TF familiale reste modeste vu la morbidité et la mortalité de la maladie, quelques études pointent du doigt une possible transmission autosomique dominante, cela remet en question certains dogmes considérés auparavant inébranlables.11, 14
L’étude génétique de la tétralogie de Fallot se focalise sur deux populations de patients, ceux atteints de la maladie dans sa forme syndromique, et ceux qui sont atteints de sa variante non syndromique.5, 11
La tétralogie de Fallot syndromique concerne 20% des cas. Comme son nom l’indique, c’est la TF entrant dans le cadre d’un syndrome génétique élargi, par exemple : le syndrome de Down (Trisomie 21), le syndrome d’Alagilles, le syndorme de DiGeorge…
La variante non syndromique englobe 80% des cas, elle peut être due à plusieurs déficits génétiques, les plus connus actuellement sont les mutations touchant le GATA4, le NKX2.5, le JAG1, et la famille des TBX. Cette rubrique se concentrera sur cette forme de la maladie et les gènes qui en sont responsables, dans le but de ne pas sortir du cadre thématique général de l’article étant donné la complexité et la diversité des syndromes génétiques qui peuvent s’associer à la TF.5, 11
Le premier gène à étudier est le « GATA4 » dont la mutation a non seulement été impliquée dans la tétralogie de Fallot familiale, mais aussi dans certains cas sporadiques pédiatriques, surtout au sein de la population chinoise, ce qui n’est pas surprenant vu que l’Asie détient la première place dans la prévalence des cardiopathies congénitales.15
En deuxième position, vient le « NKX2.5 » dont la mutation, étonnamment, ne cause pas des conséquences aussi graves que l’on peut imaginer, bien qu’il soit un gène codant un facteur de transcription spécifique essentiel pour le développement fœtal cardiaque.11 La mutation de ce gène, dont la localisation chromosomique est 5q34, est généralement associée aux troubles de conduction électrique atrio-ventriculaire.16 Ces derniers sont dus à la réduction de la densité des cellules du tissu nodal additionnée à des anomalies au sein des jonctions GAP cardiaques.16
Par la suite, on arrive à un gène fondamental de la tétralogie de Fallot, c’est le JAG-1, à localisation 20p12.2. Il code pour une molécule de signal embryologique impliquée dans le développement cardiaque.11, 16 Dans la littérature scientifique, ce gène est surtout étudié dans le cadre du syndrome d’Alagilles.11 Bien que ce dernier ne soit pas le sujet de cet article, et qu’on ait déjà dit qu’on traitera la tétralogie de Fallot dans sa forme non syndromique, il faudrait quand même se prononcer là-dessus vu que c’est un syndrome dont l’apparence phénotypique diversifiée n’altère pas son importance clinique qui réside dans les dysfonctions hépatiques et les déficits cardiaques qui encadrant la tétralogie de Fallot qui y est intégrée.17, 18 Pour faire simple, ce gène JAG1 fournit les instructions nécessaires pour synthétiser une protéine nommée JAGGED-1, ces protéines ciblent des récepteurs appelés « Notch » présents sur de différentes cellules de divers appareils et systèmes. Le signal émis par le complexe JAGGED1-Notch contrôle le développement embryonnaire de certaines catégories de cellules, spécialement celles du cœur, du foie, des yeux, des oreilles, voire même des cellules sanguines.16 C’est de là que le syndrome d’Alagilles devient clair, l’absence d’interaction JAGGED1-Notch explique les déformations phénotypiques faciales constatées chez les sujets atteints, cela explique aussi pourquoi on trouve des canaux biliaires diminués de diamètre et des malformations cardiaques évoquant très souvent la tétralogie.11, 19
La valeur scientifique de ce gène ne s’arrête pas là étant donné qu’il est, dans son état sauvage et naturel, impliqué dans la carcinogenèse de plusieurs processus tumoraux multi-organiques. Mais une minute, c’est quoi le rapport avec la tétralogie ? Les malades atteints de cardiopathies congénitales survivent désormais jusqu’à l’âge adulte grâce au développement qu’ont connu les moyens thérapeutiques. Ainsi, les sujets souffrants des mutations JAG-1 devront entamer une surveillance oncologique régulière puisque déjà, jusqu’à présent, on ne connaît pas exactement l’impact d’une telle mutation en ce qui concerne cet aspect en particulier, d’où la question qui se pose, est-ce qu’un JAG-1 mutant aurait un effet inhibiteur et réducteur sur l’aspect malin des processus tumoraux qu’il favorise, suivant la formule mathématique clamant qu’un chiffre négatif multiplié par un autre chiffre négatif donne, absolument, un résultat positif, ou bien, tout au contraire, ce genre de mutations ne mènerait qu’à prédisposer encore plus les sujets concernés au risque de développer une néoplasie via un mécanisme d’épée à double lame ? Un point d’interrogation reste maintenu, les recherches à propos de cela restent intensifiées, on attend toujours la conclusion !11, 16, 19
Un autre gène s’ajoute à la liste des suspects nucléiques responsables de la tétralogie de Fallot, c’est le gène « FOXC2 ». Avec cette nomination ressemblant à celle d’un des navires spatiaux de la NASA, ce gène, possédant le matricule chromosomique 16q24.1, reste fondamental pour le développement embryologique correct des reins, du cœur, et des vaisseaux lymphatiques.16 Bien que son impact s’élargit sur divers organes comme on peut le constater, il mérite d’être cité.
Un dernier couple de gènes à rajouter, les TBX5 et TBX1. Le TBX5, 12q24.21,16 est connu pour être la source de traduction d’une protéine T-box5 ayant le rôle de régulateur de l’activité d’autres gènes en se fixant sur des régions spécifiques de l’ADN. Durant la vie fœtale, ce gène s’engage dans la formation des tissus et des organes, particulièrement ceux du cœur et des extrémités, ce qui explique pourquoi ses mutations engendrent un syndrome bien connu, celui de Holt Oram.16 Un rare syndrome génétique associant des anomalies des extrémités supérieures et du cœur. Celles du cœur sont représentées par des malformations du septum interventriculaire et des troubles du rythme, allant de la bradycardie à la fibrillation. Cela s’explique par le rôle primaire et critique de ce gène dans la formation du septum et du circuit électrique coordonnant les contractions des chambres différentes.21, 22 Ces anomalies cardiaques sont similaires à celles résultantes de la mutation du « NKX2.5 », avec une simple différence de fréquence d’occurrence en faveur de ce dernier.11
Enfin, le « TBX1 », joue quasiment le même rôle que son jumeau « TBX5 » dans le développement embryonnaire, avec quelques distinctions notables, y compris la localisation chromosomique, la sienne étant immatriculée 22q11.21.16 On distingue deux sous types à propos des mutations affectant ce gène, celles retrouvées dans les formes syndromiques, à l’exemple du syndrome de DiGeorge, et celles décrites dans les formes non syndromiques, représentant 1% des cas. Entretemps, les implications à long terme de ce gène dans les TF non syndromiques demeurent difficiles à prédire vu sa récente identification dans la littérature.11
Les études concernant les associations génétiques responsables de la tétralogie de Fallot sont loin d’être achevées, elles perdureront pour longtemps. En guise de conclusion, les troubles d’interaction entre les facteurs de transcription importants dans la cardiogenèse jouent un rôle significatif dans l’installation de la tétralogie de Fallot, syndromique et non syndromique. Cela étaye le fait que cette maladie joue sur le volet familial, allant ainsi à contresens du dogme courant spéculant qu’elle ne n’est résultante que des mutations sporadiques, d’autant plus que plusieurs de ces mutations ont été retrouvées chez des sujets non symptomatiques faisant partie du cercle de parents et proches du premier degré des sujets réellement atteints.11, 23
Une chose est sûre, la génétique sera d’une importance déterminante et tranchante dans la compréhension et la prise en charge de cette maladie dans le futur.
Comprendre la Pathogénie de la Maladie !
La compréhension des mécanismes physiopathologiques de la tétralogie de Fallot, s’étalant de la vie fœtale jusqu’à l’âge adulte, est une condition sine qua non pour savoir gérer la pathologie sur le plan thérapeutique. C’est pour cette raison qu’on doit aborder quelques notions d’ordre embryologique afin de couvrir notre connaissance de cette maladie par un dôme d’explications bien résistant à toute sorte d’ambiguïté.
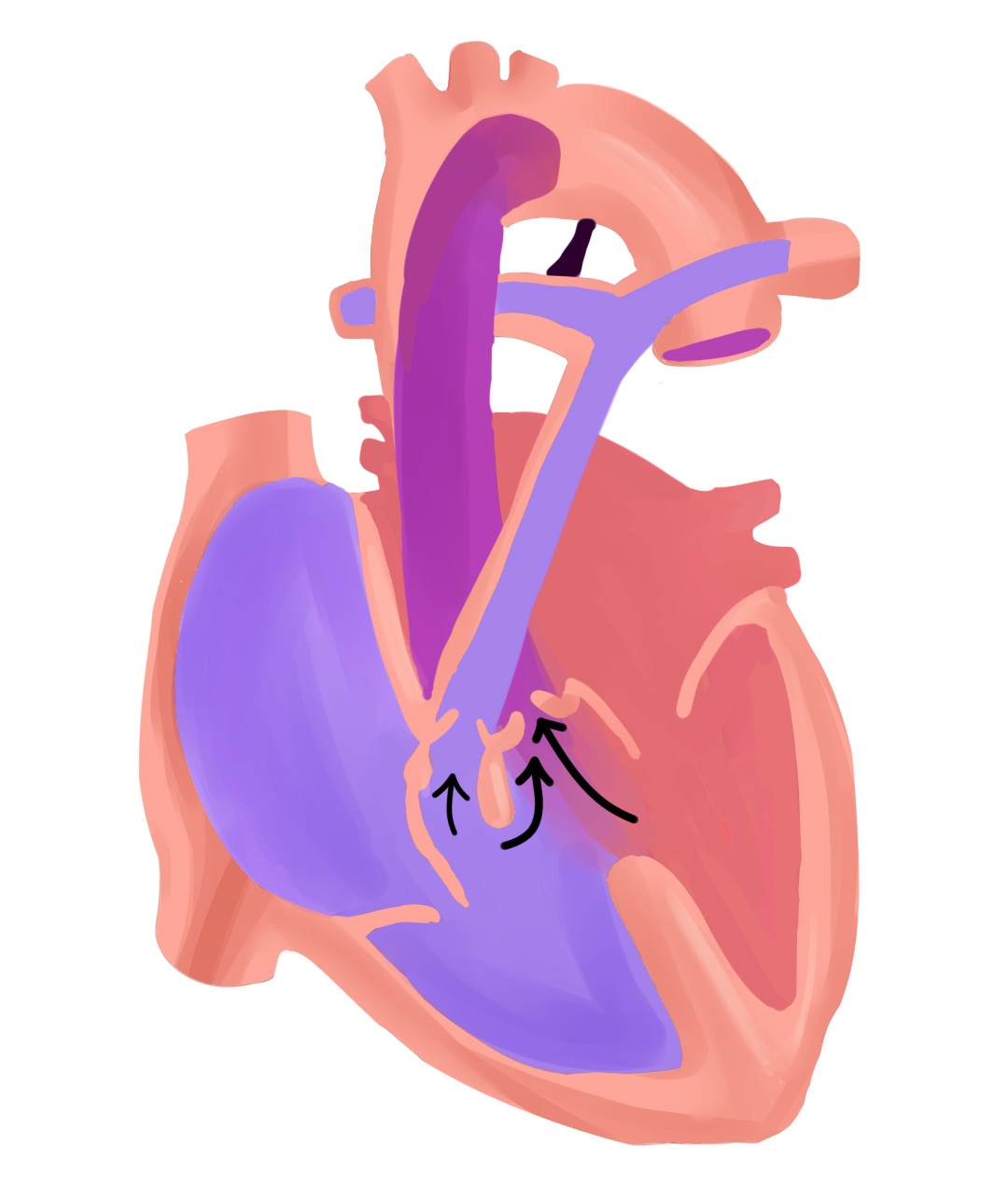
La tétralogie de Fallot résulte principalement du déplacement antérieur et supérieur, de degré variable, du septum infundibulaire, qui constitue la partie supérieure du mûr interventriculaire séparant le VD du ventricule gauche (VG).1,3 Lors du développement embryonnaire, cela provoque une division inégale du conus menant ainsi à la constitution de deux mécanismes majeurs définissant la tétralogie de Fallot.
Primo, cette division mal faite du conus entraîne un rétrécissement de l’infundibulum ventriculaire droit, la partie du VD d’où prend naissance l’artère pulmonaire via son ostium spécifique,3, 7 ce qui favorise l’installation de l’obstruction de la voie pulmonaire artérielle, premier pilier de la TF. C’est d’ailleurs ce qui cause les souffles systoliques auscultés chez l’enfant, ils sont liés au passage du sang dans le tronc pulmonaire rétréci.1, 7 Cette sténose est essentiellement musculaire,1 mais peut se compléter par la participation de l’anneau fibreux.3
En effet, la nature de la sténose suit son siège respectif, une sténose infundibulaire est généralement musculaire, elle vient en premier rang en fréquence. En deuxième place, viennent les sténoses valvulaires fibreuses, et qui sont d’ailleurs souvent associées aux sténoses infundibulaires sous-jacentes. En descendant l’échelle de fréquence encore plus bas, on retrouve les sténoses localisées au niveau des troncs artériels gauches et droits.24-26 Les sténoses constituent, en fonction de leur sévérité, un élément pronostique majeur pour les soignants.7
Le second mécanisme majeur de la maladie, étant le fameux shunt interventriculaire, est facilité par le défaut d’alignement des parois musculaires constituant le septum infundibulaire.1,3 En effet, l’attraction du septum vers le foramen pulmonaire élargit l’ouverture interventriculaire.3 Cette communication interventriculaire causée est généralement large et non restrictive.1-3, 7 C’est comme un tunnel permanent creusé et solidifié par du béton armé reliant les deux ventricules, ce qui explique pourquoi on trouve une égalisation constante des pressions entre les deux chambres ventriculaires.
Une minute ! On prend deux pas de recul, on n’a pas encore fini avec l’embryologie. On disait que le septum est attiré vers l’ostium pulmonaire causant l’obstruction de ce dernier, et que c’est par rapport au degré de cette attraction que la sévérité de la même obstruction varie.2, 7 Ce même amour, si cela se dit, que doit le septum à l’orifice met en place le troisième mécanisme majeur définissant la T4F, le chevauchement de l’aorte.
Il faut savoir que le septum, au fur et à mesure qu’il prend ses pas vers l’orifice valvulaire pulmonaire, entraîne avec lui l’aorte. Cette dernière se retrouve en train de surplomber partiellement le ventricule droit, elle le chevauche et le couvre variablement, cela est d’autant plus accentué que l’attraction du septum vers l’orifice pulmonaire est importante.24-26 Pour faire simple, c’est proportionnellement relatif à la sévérité de la T4F, allant du chevauchement négligeable dans les formes bénignes à ceux dépassant les 50% de la surface ventriculaire dans les formes sévères,1, 3 donnant ainsi ce qu’on peut nommer un VD à double issue, débouchant dans le tronc artériel pulmonaire et l’aorte. Cependant, il faut noter que la continuité entre les anneaux et les appareils valvulaires, mitral et aortique, reste préservée, la continuité fibreuse mitro-aortique est gardée intacte constamment dans la T4F.3 L’aorte ascendante peut sembler dilatée, surtout en évoluant avec l’âge, elle pourrait potentiellement engendrer une dilatation de l’anneau aortique, ce qui expliquerait une possible insuffisance aortique.24-26
Enfin, on arrive au dernier tiret de la quadruple escouade, l’hypertrophie ventriculaire droite, qui est, vraisemblablement et majoritairement, loin d’être un élément malformatif prenant naissance lors de la cardiogenèse embryonnaire comme les autres 3 piliers précédents.24 On peut constater logiquement que c’est plutôt un mécanisme réactionnel à la sténose pulmonaire oblitérant partiellement le tronc artériel, ce qui fait que l’hypertrophie est d’autant plus évidente que la sténose est sévère.3, 25, 26 Certains auteurs évoquent toutefois la possibilité que l’hypertrophie soit d’ordre congénital avec épaisseur variable de la paroi infundibulaire.3
La T4F possède un impact significatif sur l’hémodynamique corporelle, cela pourrait se résumer en trois points essentiels : la surcharge de pression du VD, l’hypoperfusion pulmonaire, et le shunt droit-gauche.
La surcharge de pression dans le VD semblerait être un phénomène perceptible car c’est la conséquence la plus logique du remplissage ventriculaire lors de la diastole en raison du blocage de la circulation provoqué par la sténose infundibulaire présente. Néanmoins, quand le gradient de pression VD-VG est assez important, l’écoulement du sang du VD au VG via la communication interventriculaire (CIV), qui est durablement large et non restrictive, fait que la pression ventriculaire droite va rapidement decrescendo pour se stabiliser dans l’intervalle des normes systémiques, indépendamment du degré de la sténose. Il est à noter que plus la sténose est serrée, plus il y aura du sang qui passera via cette communication septale pour se vider directement dans le VG, et donc dans l’aorte. Le VG, lui-même, n’est jamais sujet à des troubles de pressions.3-6
Venant à l’hypoperfusion pulmonaire, dont la relation directe avec la sténose est évidente. Elle est aussi en strict rapport avec le shunt droit-gauche qui figure comme une entité physiopathologique majeure de la maladie. Ce shunt est un peu ce qu’on expliquait tout à l’heure, le sang ne peut pas se faufiler librement et en abondance dans le tronc pulmonaire, et vu la présence de la CIV, il court-circuite, tout simplement, la petite circulation afin de se trouver dans le VG, qui fait en sorte de le propulser directement dans l’aorte. C’est le concept du VD à double issue dont on a parlé précédemment, cela pourrait certifier, de manière scientifique, la maxime anglaise qu’on vous fait avaler très souvent : «Laziness runs in my blood». Donc, attention aux courts-circuits !3, 4, 7
Le flux sanguin, en esquivant la circulation pulmonaire, est en extrême disette d’oxygène, il sera émis dans la circulation générale, ce qui mène à l’installation d’une hypoxie chronique qui a pour conséquence la cyanose caractéristique sur le plan clinique.7
Nonobstant cela, cette cyanose, inapparente chez le nouveau-né, ne devient remarquable chez le nourrisson qu’au bout de 6 mois de vie, compte tenu du fait que la sténose est modérée à la naissance et ne s’accentue qu’avec l’âge.4, 3, 24, 27
En ce qui concerne l’aspect hématologique de la maladie, la polyglobulie, sur le plan biologique, n’est que la résultante de l’hypoxie chronique, dont la manifestation clinique serait la cyanose. Cette polyglobulie s’accompagne d’une augmentation continue de l’hématocrite et de l’hémoglobine. Ces deux dernières finissent par franchir les seuils de 65% et 20g/dl respectivement, conduisant à l’apparition d’une hyperviscosité, ce qui complique au cœur la tâche d’éjecter le sang dans l’aorte, aggravant encore plus l’état d’hypoxie tissulaire, cette dernière accentuant la polyglobulie à son tour. Un cycle vicieux s’installe.3
Il ne faut pas négliger également les complications thrombotiques, particulièrement cérébrales, observées à cause de l’hyperviscosité, dont les conséquences peuvent s’avérer parfois dramatiques ! L’organisme tente de réagir en réduisant la synthèse des facteurs de coagulation. Même avec cette réponse naturelle, l’état hématologique ne demeure pas moins instable. Ainsi, on se trouve coincé dans un jeu de ping-pong entre l’hyperviscosité et ses accidents thrombotiques réccurents et la fréquente occurrence d’accidents hémorragiques provoqués par les anomalies de coagulation.3, 24, 27
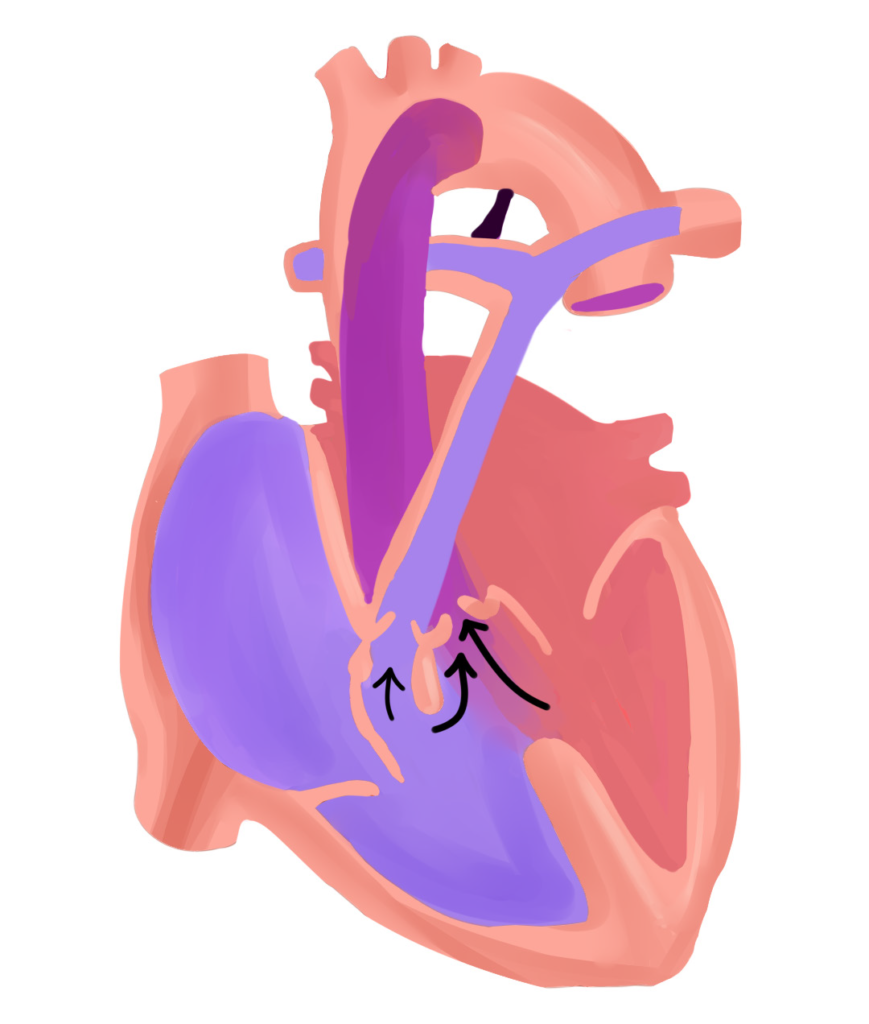
Adaptée de : Anderson RH, Penny D, et al: Paediatric Cardiology, ed 3, 2009, Page 330.
Reproduction de Meriem AFIR
Comment Diagnostiquer une Tétralogie de Fallot ?
Comme la vaste majorité des cardiopathies congénitales, la tétralogie de Fallot est, de nos jours, diagnostiquée lors de la période fœtale grâce à l’avancement technologique.1 Pour aborder la clinique, il faut déjà rappeler que la tétralogie de Fallot ne s’accompagne généralement d’aucun signe apparent à la naissance étant donné que la sténose est plus ou moins minime, ce qui fait qu’il n y a toujours pas de shunt droit-gauche important, et donc la circulation pulmonaire reste quasi intacte. On peut, toutefois, noter un souffle systolique.1 Ce n’est que lorsque la sténose devient considérable ou dès que les résistances pulmonaires augmentent que la cyanose s’observe graduellement. Cela survient lors des six premiers mois de vie, et la probabilité de faire une crise hypoxique devient de plus en plus significative.
Comment apparaissent ces crises ? Il aurait été possible de les étaler dans le chapitre de physiopathologie, mais ce n’est jamais trop tard ! En effet, elles sont en rapport avec l’oblitération subtotale à totale du tronc artériel pulmonaire par aggravation paroxystique de la sténose en cause. Ainsi, les crises se déclenchent inopinément à la faveur de divers facteurs déclencheurs, tels que l’effort, l’agitation, ou bien même la déshydratation, mais, encore une fois, comment cela se produit-il ? Cela se joue sur les cordes de pression, par exemple lors d’une diminution des résistances systémiques par une chute de pression engendrée par l’hypovolémie en cas de déshydratation, ou bien lors d’une augmentation des résistances vasculaires pulmonaires par un spasme provoqué par la noradrénaline sécrétée en exerçant l’effort, quel que soit le cas, l’éjection sanguine dans l’aorte sera favorisée dans le but d’augmenter le débit cardiaque, et donc le shunt droit-gauche sera renforcé. Par conséquent, un flux sanguin artériel désoxygéné plus important est pompé dans la circulation générale.3, 4, 7
Un autre signe doit être évoqué, c’est le «squatting», qui consiste en une attitude physique permettant au malade de mieux gérer l’hypoxie, comment cela se passe-t-il ? L’accroupissement engendre une augmentation des résistances périphériques suivie d’une diminution du shunt droit-gauche, ce qui favorise une meilleure éjection sanguine dans la circulation pulmonaire.
Un dernier signe fonctionnel s’ajoute à la liste, c’est l’hyperpnée. Elle provoque une dépression thoracique, qui, en favorisant le retour veineux, induit une surcharge ventriculaire en regard de la sténose infundibulaire, le shunt droit-gauche s’aggrave alors sous l’effet de l’excès de pression, ce qui, non seulement renforce l’état d’hypoxie, mais mène aussi à l’apparition d’une hypercapnie surajoutée et l’installation d’une acidose. Tous ces phénomènes favorisent à leur tour la prolongation de l’hyperpnée, qui les a causés, dans l’espoir d’effectuer une compensation, sauf que cela ne fait que noyer l’organisme dans un cycle vicieux rétro-actif, accentuant sa gravité avec la persistance de l’hyperpnée.3, 29
On retrouve également, comme signes cliniques, l’hippocratisme digital et le retard staturo-pondéral.3, 7
L’imagerie, véritable clé du diagnostic :
En arrivant à la partie des examens complémentaires, on trouve une gamme variée permettant d’identifier et de confirmer le diagnostic de la maladie. En première position vient la radiographie thoracique dont on ne peut se passer. Qui dit radiographie thoracique de la tétralogie de Fallot, dit cœur en sabot, hautement spécifique de la pathologie. Cet aspect est caractérisé par une pointe surélevée, signe de HVD, un arc moyen concave, signe d’hypoplasie du tronc pulmonaire, et un bouton aortique proéminent, reflétant une dilatation de l’aorte, généralement présente, comme sus-mentionné. Il s’ajoute à cela des signes d’hypo-perfusion pulmonaire.3, 7, 24, 29
Un autre examen s’offre au diagnostic paraclinique de la maladie, c’est l’échocardiographie transthoracique, qui est d’une importance incontestable, étant donné qu’elle permet la visualisation du cœur et de son anatomie interne de différents angles standards,4, 7 chacun ayant son intérêt spécifique, que cela soit dans la localisation et l’appréciation de l’extension ou même dans la quantification du nombre de trous existants au niveau du septum, sans oublier la détection des anomalies valvulaires. Les différents axes de vue portent un coup de main dans l’évaluation méticuleuse des caractéristiques anatomiques définissant la tétralogie de Fallot, certains sont d’une importance considérable. Le doppler, appoint non-négligeable à cet examen d’imagerie, permet d’estimer le gradient de pression tout au long de la voie pulmonaire, ce qui renforce l’acuité de l’évaluation.3, 4, 7, 31
D’autres examens plus avancés s’additionnent à la gamme d’examens d’exploration. L’IRM et la TDM, en première place, attirent de plus en plus d’attention en devenant le gold standard en ce qui concerne l’évaluation des cardiopathies congénitales, plus particulièrement dans les pays où les moyens technologiques sont aisément à la portée des établissements hospitaliers.7
En matière de tétralogie de Fallot, l’IRM est moins fréquemment indiquée en pré-opératoire puisque la phase échocardiographique rapporte suffisamment de données nécessaires. Cependant, c’est inversement le cas quand il s’agit de l’évaluation post-opératoire, l’IRM permet une optimisation exemplaire de l’estimation quantitative du volume ventriculaire droit et de ses fonctions, de la fonctionnalité des valves pulmonaires, ainsi que la détection des potentielles anomalies du VG.7, 30, 32, 33 Elle permet également de mesurer le diamètre des troncs artériels, et de révéler, avec précision, non seulement les sténoses qui y sont présentes, mais aussi la distribution du flux sanguin entre les artères pulmonaires, droite et gauche.7
La TDM, avec ses capacités phénoménales de mappage spatio-temporel à haute résolution, demeure encore une meilleure option dans l’évaluation des aspects anatomiques de la T4F, que cela soit en pré- ou postopératoire, surtout s’il y a une difficulté de visualisation par échocardiographie, ou en cas de contre-indications à l’usage de l’IRM. Son rôle est également prometteur dans le suivi évaluatif des cardiopathies congénitales en général.7
La cathétérisation cardiaque, par contre, n’est pas d’usage de routine quand il s’agit d’une tétralogie de Fallot classique vu l’existence d’outils assez performants et moins invasifs, tels que l’IRM et le Pet-Scan, surtout que les approches par cathéter peuvent parfois déclencher des crises hypoxiques, elles doivent être menées avec prudence. Parfois on n’a pas le choix, spécialement quand c’est d’intérêt thérapeutique, dans le but de dilater des artères pulmonaires par des ballons gonflables, de poser des stents, ou bien même d’intervenir sur des vaisseaux collatéraux endommagés. Dans tous les cas, la balance bénéfice/risque doit être évaluée minutieusement.5, 7, 34, 35
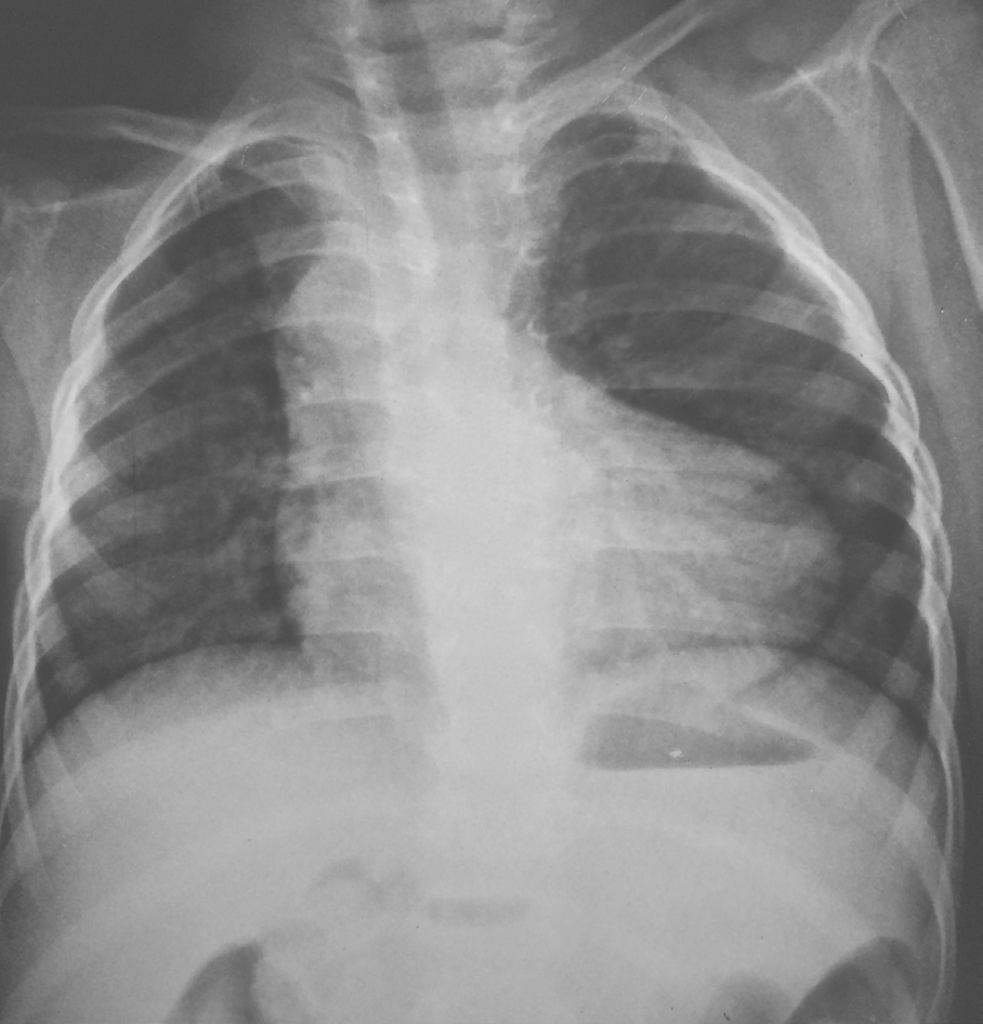
caractéristique de la maladie de tétralogie de Fallot chez un enfant de 5 ans.
Tirée de : Case courtesy of Dr Vincent Tatco, Radiopaedia.org, rID: 43227
La Tétralogie de Fallot, Comment la Gère-t-on ?
À l’époque, 50% des sujets atteints décédaient dès les premiers jours de vie, bien avant qu’un réel plan d’action thérapeutique curatif ne soit tracé.2, 36 Désormais, c’est la chirurgie à cœur ouvert qui sauve ces vies, à tel point que les patients parviennent à survivre jusqu’à l’âge adulte.2 Les stratégies actuelles de traitement sont variées, mais, en général, elles se fondent sur deux volets principaux, médical et chirurgical.
Le traitement médical, qui est dans tous les cas palliatif, consiste en la correction du taux de fer sanguin en cas de présence d’anémie ferriprive, ainsi que la prise en charge urgente des crises hypoxiques par l’administration de l’oxygène en position genu-pectorale, en pliant les genoux sur l’abdomen, associée à l’injection de propranolol par voie IV. Si une hémorragie ou une hypotension apparaît dans le contexte de la crise, l’exécution a priori d’un remplissage plasmatique et/ou une transfusion sanguine demeurent une réflexion de base.3
En passant au volet chirurgical, on doit préciser qu’on y distingue deux variantes, la chirurgie palliative et la chirurgie curative, l’opérabilité représente de facto l’élément sur lequel on se base pour choisir quelle voie engager. On explique !
La première option, étant la chirurgie à cœur ouvert vu que c’est l’approche chirurgicale curative primordiale de la maladie, est effectuée prioritairement par la majorité des spécialistes.3 L’âge de la pratique de cette chirurgie s’est graduellement réduit depuis sa première élaboration en 1955,2, 37 une idée répandue présume que plus tôt l’intervention se fait, plus l’exposition à la surcharge de la pression ventriculaire droite et à la désaturation d’oxygène sera limitée, ce qui préserve mieux les fonctions cardiovasculaires et cérébrales des risques ischémiques.38-40 La majorité des centres spécialisés préfèrent la retarder jusqu’à un intervalle d’âge approximatif entre les 3 à 6 mois de vie étant donné que les opérations en stade néonatal, autrement dit, avant de clôturer le 1 mois de vie, n’ont pas démontré de meilleurs résultats à court terme que ceux des opérations se procurant au-delà de l’âge néonatal.38, 39
Le chirurgien a la possibilité d’effectuer la réparation par plusieurs méthodes. La première est de gérer la sténose du tronc pulmonaire par une ventriculotomie via le mur ventriculaire antérieur, suivie d’une résection extensive du segment d’émergence du tronc pulmonaire, la partie qui relie le VD au tronc pulmonaire, et l’installation d’un patch trans-annulaire de remplacement. À savoir que c’est celle-ci qui est communément pratiquée, elle est, toutefois, généralement associée à une physiologie ventriculaire droite restrictive comme complication à court terme. La deuxième méthode est formée de la même technique, juste en épargnant la valve pulmonaire, au prix de laisser encore plus de lésions résiduelles obstructives non-réséquées.1, 3
Le chirurgien se trouve parfois forcé de pratiquer ces opérations en âge néonatal chez les sujets précocement symptomatiques, surtout ceux qui se présentent avec des crises hypercyanotiques.2 Dans le cas où ces interventions, d’ordre primaire, ne sont pas tolérées, ou que le bilan d’opérabilité est défavorable vis-à-vis de leur faisabilité, une mise en place d’un plan d’action de second ordre est cruciale, d’autres stratégies thérapeutiques sont abordées !
C’est de là que s’identifie la chirurgie palliative qui représente une solution temporaire jusqu’à ce que l’état général du sujet lui permette d’effectuer l’opération curative définitive. Bien que les indications de la chirurgie palliative soient précises et limitées, la plupart des chirurgiens préfèrent pratiquer une réparation d’emblée, notamment chez les nouveau-nés.3 La chirurgie palliative figure comme un moyen d’augmenter le flux pulmonaire, de réduire l’hypoxie, et de développer le lit vasculaire, ou du moins le préserver. Elle est assurée par la création d’un shunt systémico-pulmonaire en suivant l’une de deux procédures connues : (1) soit en effectuant l’intervention de Blalock-Taussig par la réalisation d’une anastomose entre l’artère sous-clavière et l’artère pulmonaire homolatérale, cela risque de priver le bras de son vaisseau d’afflux sanguin artériel principal, ce qui est toléré chez le nourrisson, surtout que ce n’est que pour une durée limitée ; (2) soit via l’anastomose de Blalock-Taussig modifiée, qui consiste en l’installation d’un stent reliant les deux artères précédentes, ce qui permet un meilleur calibrage en fonction de l’âge et du poids du sujet.3, 38, 41
Dès que les conditions s’y prêtent, on rappelle le patient pour l’opération curative.
D’autres stratégies thérapeutiques sont mises en œuvre, telle que la dilatation au ballonnet du segment d’émergence du tronc pulmonaire par cathétérisme. On rappelle que ce genre d’intervention présente ses propres risques consistant en un déclenchement de crises hypoxiques, et donc doit être utilisé avec soin par des spécialistes. Néanmoins, cela ne diminue en rien son intérêt primordial, notamment dans le soulagement des sténoses pulmonaires et l’amélioration de la saturation des globules rouges en oxygène.3
Conclusion
Jadis, la tétralogie de Fallot était un fléau. À peine né, le nouvel arrivé au monde se trouvait avec une peine de mort sur les épaules, la malchance !
Heureusement, de nos jours, notre connaissance sur cette maladie s’est approfondie, elle s’est multipliée. Les recherches par rapport aux cardiopathies congénitales sont toujours d’actualité, on ne risque pas de tomber à sec de nouveautés, on comprend de mieux en mieux les complications à long terme que les patients post-opérés pourraient développer, cela se reflète sur la qualité de leurs suivi et prise en charge qui ne cessent de s’améliorer.
Une chose est sûre, la bataille contre la tétralogie de Fallot a tracé, au fil du temps, un parcours honorable, lustré de gloire. Ainsi, s’annonça n’avoir jamais été plus proche, l’heure de la grande victoire !
Références
1- Tom R. Karl, MS, MD, FRACS; Christian Stocker, MD, FRACP. Tetralogy of Fallot and its Variants, 2016.
2- Christian Apitz, Gary D Webb, Andrew N Redington. Tetralogy of Fallot, The Lancet, 2009.
3- M. KHAYAT Ali. LA CHIRURGIE DE LA TETRALOGIE DE FALLOT, Thèse N° 095/18, 2018.
4- Pooja Swamy, M.D., Aditya Bharadwaj, M.D., Padmini Varadarajan, M.D., and Ramdas G. Pai, M.D. Echocardiographic Evaluation of Tetralogy of Fallot*, 2014.
5- Lisa Wise‐Faberowski, Ritu Asija, Doff B. McElhinney. Tetralogy of Fallot: Everything you wanted to know but were afraid to ask, 2018.
6- Michael Ma, Richard D. Mainwaring, and Frank L. Hanley. Comprehensive Management of Major Aortopulmonary Collaterals in the Repair of Tetralogy of Fallot, 2017.
7- Christian Apitz, Roboert H. Anderson, Lynn Dees, James S. Tweddell, Diane E. Spicer, Andrew N . Redington. Tetralogy of Fallot with Pulmonary Stenosis, Anderson’s Pediatric Cardiology p2388-p2453.
8- Lev M, Eckner FAO. The pathologic anatomy of tetralogy of fallot and its variants. Dis Chest. 1961;45:251–261.
9- Stenson N. Embrio monstro affinis parisiis dissectum. Acta Med Philos Hafniensa. 1671;72(1):202–20.
10- Hunter W. Medical observations and inquiries. London: Private publication, 1784: 417–19.
11- Ari Morgenthau MD, William H. Frishman MD, Genetic Origins of Tetralogy of Fallot.
12- Burn J, Brennan P, Little J, et al. Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study. Lancet 1998;351: 311- 316.
13 – Chen-Yee NJ, Costain-G, swaby J-A, et al. Reproductive fitness and genetic transmission of tetralogy of Fallot in the molecular age. Circulation: Cardiovascular genetics 2014;7:102-109.
14- Di Felice V, Zummo G. Tetralogy of fallot as a model to study cardiac progenitor cell migration and differentiation during heart development. Trends Cardiovasc Med 2009;19: 130-135.
15- Zhang W, Li X, Shen A, et al. GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur J Med Genet 2008;51: 527-535.
16- U.S National Library of Medicine, Genetics Home Reference. https://ghr.nlm.nih.gov.
17- Alagille D, Estrada A, Hadchouel M, et al. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediat 1987;110: 195-200.
18- Emerick KM, Rand EB, Goldmuntz E, et al. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999;29: 822-829.
19- Robert C Bauer & al. Jagged1 (JAG1) Mutations in Patients With Tetralogy of Fallot or Pulmonic Stenosis. 2010 May;31(5):594-601.
20- Fang J, Dagenais SL, Erickson RP, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet 2000;67: 1382-1388.
21- Wilson V, Conlon FL. The T-box family. Genome Biol 2002;3: 3008.
22- Basson CT, Bachinsky DR, Lin RC, et al., Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 1997;15:30-35.
23- Gatzoulis MA, Balaji S, Webber SA, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of Tetralogy of Fallot: a multicentre study. Lancet 2000; 356: 975-981.
24- ISELIN M. Cardiopathies congénitales. Encycl Méd Chir, Radiodiagnostic – Cœur-Poumon 32-015-A-12, Pédiatrie, 4-070-A-05, 1999, 6 p.
25- CHAUVAUD S. Tétralogie de Fallot : généralités. Encycl Méd Chir, Techniques Chirurgicales – Thorax, 42-800, 2003, 7 p.
26- FRIEDLI B. Tétralogie de Fallot. Encycl Méd Chir, Cardiologie, 11-940-E-50, 2002, 9 p.
27- HEITZ F. Cardiopathies congénitales. Encycl Méd Chir, AKOS Encyclopédie Pratique de Médecine, 8-0680, 1998, 14 p.
28- Carvalho JS. Fetal heart scanning in the first trimester. Prenatal diagnosis. 2004;24(13):1060-7.
29- FRIEDLI B. Tétralogie de Fallot. EMC-Pédiatrie 1 (2004) 365–378.
30- AHA recommendations, Tetralogy of Fallot, e749.
31- Snider AR, Silverman NH: Suprasternal notch echocardiography: A two-dimensional technique for evaluating congenital heart disease. Circulation 1981;63: 165–173.
32- Helbing WA, Rebergen SA, Maliepaard C, et al. Quantification of right ventricular function with magnetic resonance imaging in children with normal hearts and with congenital heart disease. Am Heart J. 1995;130:828–837.
33- Rebergen SA, deRoos A. Congenital heart disease. Evaluation of anatomy and function by MRI. Herz. 2000;25:365–383.
34- Kohli V, Azad S, Sachdev MS, et al. Balloon dilation of the pulmonary valve in premature infants with tetralogy of Fallot. Pediatr Cardiol. 2008;29:946‐959.
35- Jonas RA. Tetralogy of Fallot with pulmonary atresia. In: Jonas RA, ed. Comprehensive Surgical Management of Congenital Heart Disease, 2nd edn. Boca Raton, FL: CRC Press, Taylor & Francis Group; 2014:585‐603.
36- Bertranou EG, Blackstone EH, Hazelrig JB, Turner ME, Kirklin JW. Life expectancy without surgery in tetralogy of Fallot. Am J Cardiol 1978; 42: 458–66.
37- Lillehei CW, Cohen M, Warden HE, et al. Direct vision intracardiac surgical correction of the tetralogy of Fallot, pentalogy of Fallot, and pulmonary atresia defects; report of fifi rst ten cases. Ann Surg 1955; 142: 418–42.
38- Jelle P.G. van der Ven, Eva van den Bosch, Ad J.C.C. Bogers, Willem A. Helbing. Current outcomes and treatment of tetralogy of Fallot.
39- Loomba RS, Buelow MW, Woods RK: Complete Repair of Tetralogy of Fallot in the Neonatal Versus Non-neonatal Period: A Meta-analysis. Pediatr Cardiol. 2017; 38(5): 893–901.
40- Daliento L, Mapelli D, Russo G, et al.: Health related quality of life in adults with repaired tetralogy of Fallot: Psychosocial and cognitive outcomes. Heart. 2005; 91(2): 213–8.
41- Kiran U, Aggarwal S, Choudhary A, et al.: The blalock and taussig shunt revisited. Ann Card Anaesth. 2017; 20(3): 323–330.