
Hypothyroïdie Congénitale – Mise au Point
Hadya LAGGOUN
L’hypothyroïdie congénitale (HC) est l’anomalie congénitale endocrinienne la plus fréquente dans le monde, avec une incidence qui a quasiment doublé ces dernières années. Elle est l’une des causes les plus courantes du retard mental dans le monde, fort heureusement évitable si celle-ci est traitée dans les 2 premières semaines de vie. La plupart des nouveau-nés souffrant d’hypothyroïdie congénitale ont cependant peu ou pas de manifestations cliniques signant une carence en hormones thyroïdiennes. De plus, la majorité des cas sont sporadiques, de sorte qu’il n’est pas possible de prédire quels nourrissons sont susceptibles d’être atteints. Quel est donc le moyen qui a été mis au point et qui permet de détecter une HC le plus tôt possible chez un nouveau-né ? Quelle serait son implication dans l’augmentation de l’incidence de cette affection ? Cet article vise à répondre à ces questions tout en fournissant un résumé des progrès récents et significatifs dans l’étiopathogénie de l’hypothyroïdie congénitale. Ces derniers pourraient ouvrir de nouvelles pistes vers un diagnostic anténatal et éventuellement un traitement in utero de l’HC.
Pendant la grossesse, l’unité mère-fœtus doit produire environ 40% de plus d’hormones thyroïdiennes pour répondre de manière adéquate aux besoins de la mère et du fœtus en développement.1 Un apport insuffisant à l’organisme en ces hormones caractérise l’hypothyroïdie congénitale. En raison du rôle essentiel joué par les hormones thyroïdiennes dans la croissance et le développement du cerveau in utero et après la naissance2 (figure), si elle n’est pas traitée immédiatement, l’HC peut entraîner un retard de croissance et des troubles neurologiques et psychiatriques ; notamment une déficience intellectuelle irréversible qui est la caractéristique clinique la plus pertinente et la plus dévastatrice de l’HC.3 En effet, il existe une relation inverse entre l’âge d’initiation du traitement et le quotient intellectuel (QI) et chaque jour de retard pourrait entraîner une perte du QI, d’autant plus que le nouveau-né est plus jeune.4 L’HC peut être permanente, nécessitant un traitement substitutif à vie, ou transitoire désignant une déficience temporaire redevenant euthyroïdienne au cours des premiers mois ou des premières années de vie.
Pathogénie de l’hypothyroïdie congénitale permanente
L’hypothyroïdie congénitale permanente d’origine centrale (hypophysaire ou hypothalamique) est très rare.
Elle survient de manière isolée (déficit isolé en thyrotropine : TSH – Thyroid Stimulating Hormone -) ou le plus souvent combinée à d’autres déficits hormonaux hypophysaires. Les mécanismes moléculaires sous-jacents à l’HC centrale sont encore largement méconnus. À l’heure actuelle, une déficience isolée en TSH a été associée à des mutations inactivatrices de quatre gènes : TSH-beta (TSH beta-subunit), TRH-R (Thyrotropin-Releasing Hormone Receptor), IGSF1 (Immunoglobulin Superfamily Member 1) et TBL1X (Transducin Beta-Like protein 1X).5
La forme permanente la plus courante de cette endocrinopathie est l’HC primaire, caractérisée par des taux élevés de TSH reflétant une anomalie du développement de la glande thyroïdienne (dysgénésie) ou un trouble de l’hormonosynthèse.
La dysgénésie thyroïdienne (DT) représente 80 à 85% de tous les cas d’HC primaire. Elle constitue un groupe hétérogène d’affections résultant de la perturbation d’une des étapes du développement de la thyroïde, allant de la génération de thyrocytes fonctionnels à partir de cellules souches pluripotentes endodermiques à la migration de la thyroïde du foramen caecum, jusqu’à sa position basi-cervicale antérieure définitive. La DT comprend ainsi l’agénésie thyroïdienne ou l’athyréose, l’hypoplasie d’une glande orthotopique et l’ectopie thyroïdienne. L’agénésie thyroïdienne, forme la plus grave de DT, se caractérise par les taux sériques de TSH les plus élevés et par des taux de thyroglobuline (TG) sérique indétectables. On ignore encore si l’athyréose découle de l’arrêt de différenciation des cellules endodermiques pluripotentes ou de l’apoptose du pool de précurseurs des thyrocytes au cours des premières étapes de l’organogenèse. L’ectopie thyroïdienne est la forme la plus fréquente de DT. Elle est associée à un dysfonctionnement thyroïdien variable : les nouveau-nés affectés peuvent présenter une hypothyroïdie grave au moment de la naissance, voire même une hypothyroïdie à révélation tardive qui échappe à un diagnostic néonatal.6
Bien que la plupart des cas d’HC avec dysgénésie se présentent sous forme sporadique, de plus en plus de preuves indiquent que des facteurs génétiques y sont impliqués. Ceci inclut l’observation d’une prévalence féminine significative pour l’ectopie mais pas pour l’athyréose7 et d’une différence d’incidence de DT selon les groupes ethniques (c-à-d. une susceptibilité plus élevée chez les enfants asiatiques d’origine indienne, hispaniques et inférieure chez les enfants noirs).8 De plus, l’incidence de l’HC est plus élevée dans les populations où le nombre de mariages consanguins est élevé.9 L’association dans 5 à 6% des cas de DT à d’autres malformations congénitales majeures corrobore également l’hypothèse de l’origine génétique de la DT.10 Enfin, les mutations dans les gènes impliqués dans le développement de la thyroïde provoquent des DT dans des modèles animaux.11
Néanmoins, la DT a été trouvée discordante chez les jumeaux monozygotes12 et l’analyse de liaison et des mutations de cas familiaux a démontré une hétérogénéité génétique.13 En outre, un modèle murin a démontré l’origine multigénique de la DT chez la souris.14 Toutes ces données permettent de suggérer qu’il existe un spectre large de DT allant des DT de causes monogéniques aux DT multifactorielles, où les facteurs environnementaux et épigénétiques semblent impliqués. A ce jour, de rares cas de DT ont été associés à des mutations de gènes codant d’une part pour des facteurs de transcription impliqués dans le développement de la thyroïde (PAX-8, NKX2.1, FOXE1 et NKX2.5) ; et d’autre part pour le récepteur TSH (TSHR) et le gène GS-alpha (GNAS).15-18 Des mutations des gènes GLIS3, jagged 1 (JAG1) et borealin (CDCA8) peuvent également contribuer à la pathogenèse de la DT.19-21
Les 15% de cas restants d’hypothyroïdie congénitale permanente sont dus à un trouble de l’hormonosynthèse qui est souvent détecté à la naissance. La dyshormonogenèse thyroïdienne est causée par des mutations de gènes impliqués dans l’une des étapes de la synthèse des hormones thyroïdiennes à savoir le transport de l’iode dans la cellule folliculaire (NIS : Symporteur Sodium Iodure) et vers la colloïde (PDS : Pendrine), l’oxydation de l’iode (TPO : Thyroperoxydase), la génération de H2O2 (THOX2 : Thyroïde Oxydase de type 2), la synthèse de la thyroglobuline (TG) et le recyclage de l’iodine par voie intrathyroïdienne (IYD).22
L’hypothyroïdie congénitale périphérique est une catégorie distincte de l’HC permanente. Elle résulte d’anomalies du transport des hormones thyroïdiennes à travers la membrane cellulaire (mutations dans le transporteur de monocarboxylate 8, MCT8), de leur métabolisme (mutations dans la protéine intervenant dans la synthèse des sélénoprotéines 2, SBP2) ou encore de leur action nucléaire (mutations du récepteur de l’hormone thyroïdienne alpha et les gènes béta, THRA et THRB).23
Pathogénie de l’hypothyroïdie congénitale transitoire
La prévalence d’HC transitoire varie selon le statut en iode : elle est plus élevée dans les zones où la carence en iode est plus sévère.24 La surcharge en iode pendant la période périnatale est également une cause d’HC transitoire par effet Wolff-Chaikoff.25 Cela peut se produire chez les nourrissons de mères atteintes d’arythmie cardiaque traitée à l’amiodarone, ou lorsque des composés antiseptiques contenant de l’iode sont utilisés chez la mère ou le nourrisson. L’exposition transitoire de femmes enceintes à un agent de contraste iodé dans le cadre d’une tomodensitométrie ne semble pas avoir d’effet sur la fonction thyroïdienne néonatale.26
L’HC transitoire a été associée à un transfert transplacentaire de médicaments antithyroïdiens utilisés pour traiter l’hyperthyroïdie maternelle ; dans ce cas, l’hypothyroïdie dure quelques jours à 2 semaines après la naissance. Aussi, un transfert transplacentaire d’anticorps bloquant les récepteurs de TSH (Ac anti RTSH) peut survenir chez les nourrissons de mères atteintes d’une maladie thyroïdienne auto-immune.27 Un tel diagnostic doit alors être envisagé si plus d’un nourrisson né de la même mère est identifié comme ayant une hypothyroïdie primaire par le dépistage néonatal. Cette forme d’hypothyroïdie disparaît généralement en un à 3 mois à mesure que les anticorps maternels sont éliminés.
En outre, on a signalé que les grands hémangiomes hépatiques, présents dès la naissance, peuvent entraîner une augmentation des taux de désiodase de type 3 induisant ainsi une hypothyroïdie dite de consommation. Celle-ci disparaît après le traitement de la tumeur.28
Enfin, des mutations du gène DUOX2 intervenant dans la génération de peroxyde d’hydrogène indispensable à la peroxydase thyroïdienne dans la synthèse d’hormones thyroïdiennes, peuvent expliquer de rares cas d’HC transitoires.29 Toutefois, comme le montre une étude récente,30 tous les facteurs de risque énumérés ci-dessus ne peuvent expliquer qu’une petite fraction des cas d’HC transitoire, tandis que la plupart restent inexpliqués.
Comme il peut être difficile au cours de la période néonatale de différencier l’hypothyroïdie permanente de l’hypothyroïdie transitoire, un traitement substitutif par la lévothyroxine (L-T4) doit être instauré chez tous les nouveau-nés avec un test de dépistage positif, et ce sans attendre les résultats de confirmation du diagnostic d’HC. Pareillement, les autres études de diagnostic (scintigraphie et échographie, par exemple) visant à déterminer la cause sous-jacente de l’HC ne devraient pas retarder le début du traitement. Dans ce cas, une réévaluation diagnostique doit être effectuée à l’âge de 3 ans afin de faire la distinction entre une HC permanente et transitoire. Elle peut être anticipée à la fin de la première ou de la deuxième année de vie chez les nourrissons avec une suspicion élevée d’HC transitoire.
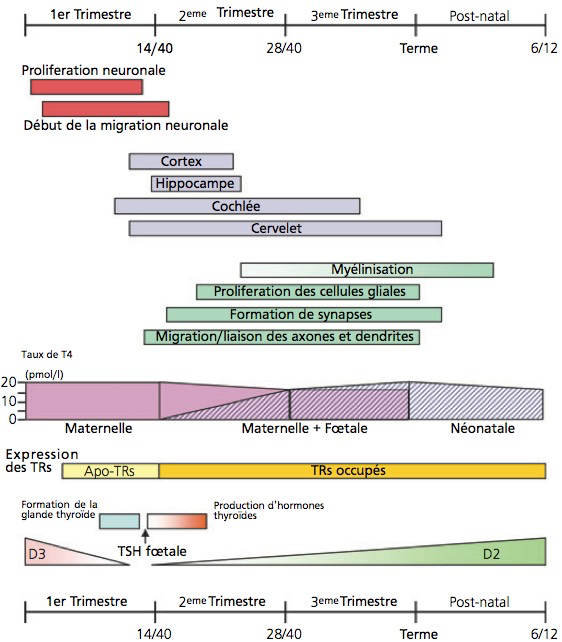
Tiré de Williams G. R. (2008). Neurodevelopmental and neurophysiological actions of thyroid hormone. J. Neuroendocrinol. 20, 784–794.
Dépistage de l’hypothyroïdie congénitale
À la naissance, seulement 1 à 4 % des cas d’HC sont diagnostiqués cliniquement. La sémiologie comporte une peau sèche, marbrée, un ictère néonatal prolongé, une macroglossie, une hypothermie, une léthargie, une hypotonie, des difficultés à la succion, une hernie ombilicale et une constipation, surtout si l’enfant est nourri au sein.31 La persistance de la fontanelle postérieure, d’une grande fontanelle antérieure et d’une large suture sagittale peuvent être d’autres signes de la maladie reflétant un retard de maturation osseuse. Quelques nourrissons atteints de dyshormonogenèse thyroïdienne peuvent également présenter un goitre palpable, un goitre et une surdité dans le cas du syndrome de Pendred. La plupart de ces manifestations ne sont pas présentes à la naissance et les caractéristiques cliniques sont souvent initialement subtiles et non spécifiques. Ceci est dû d’une part à l’effet protecteur de l’hormone thyroïdienne maternelle, qui traverse le placenta jusqu’à la fin de la grossesse ; et d’autre part à la présence d’un tissu thyroïdien fonctionnel dans la forme la plus courante d’HC. Si l’HC reste non traitée, les manifestations cliniques deviennent plus évidentes au cours de la seconde moitié de la première année de vie, marquées par un retard de croissance, un retard dans le développement des habiletés motrices et une déficience intellectuelle.
L’introduction du dépistage néonatal au milieu des années 70, à la suite du travail pionnier de J. Dussault au Québec,32 a rendu possible le diagnostic précoce de l’hypothyroïdie congénitale. Des controverses subsistent quant au programme de dépistage optimal et à la gestion de ce trouble. Trois procédures majeures de dépistage ont été suivies33 : une méthode reposant sur un dosage primaire de la thyroxine libre (T4l) puis de la thyrotropine (TSH) chez les nourrissons dont la valeur de T4l est inférieure à un seuil choisi ; une autre reposant sur le dosage primaire de la TSH et une dernière méthode fondée sur des dosages simultanés de T4 et de TSH. Le dépistage est effectué sur papier buvard sur lequel sont disposées des gouttes de sang du nouveau-né par prélèvement capillaire au talon, et ce dans les premiers jours suivant la naissance. La meilleure fenêtre pour le test va de 48 à 72 heures après la naissance, car une mesure précoce de la TSH a une fréquence élevée de résultats faussement positifs en raison de la poussée physiologique néonatale de TSH.
Chaque approche présente des avantages et des inconvénients. Le dosage primaire de la T4 a une faible spécificité car l’hypothyroxinémie est fréquente chez les prématurés et dans les anomalies congénitales de la Thyroid Binding Globulin (TBG). Néanmoins, les nourrissons présentant une élévation retardée de la concentration sanguine de TSH34 et ceux présentant une hypothyroïdie centrale sont détectés de manière plus fiable par cette méthode. Quant à l’HC primaire, le dosage primaire de la TSH est le test le plus sensible pour détecter même la forme subclinique de celle-ci (TSH sanguine élevée, T4 sanguin normal). Effectivement, les taux sériques de TSH ainsi que le log TSH sont inversement proportionnels à la concentration en FT4 ; par conséquent, de légères variations dans les taux de la T4l sont reflétés par des changements importants dans les taux de la TSH sérique. Enfin, aucun des programmes précédents ne permet de détecter les nourrissons présentant une HC périphérique. C’est dans ce but que des programmes pilotes, mesurant à la fois la T4 et la TSH, ont été entrepris dans certains pays.35 Le nombre de faux positifs, extrêmement faible, peut être lié à une forme transitoire d’HC, une carence iodée modérée, une prématurité ou à une erreur de dosage. Quelques cas de faux négatifs ont été rapportés, incitant à surveiller les signes cliniques pouvant faire suspecter la maladie et à demander un dosage hormonal en cas de doute.
Des seuils pour les résultats des tests sont définis pour chaque programme de dépistage néonatal. En règle générale, si la T4 de dépistage est inférieure au 10ème percentile et/ou si la TSH est supérieure à la limite spécifique du centre de dépistage, le nourrisson est rappelé pour confirmation du diagnostic. Il est nécessaire de savoir que les résultats obtenus sur éluats sur papier buvard n’ont pas de valeur diagnostique. Les valeurs sont au minimum semi-quantitatives et aident à l’identification des individus potentiellement atteints d’une HC. Tout résultat anormal doit être confirmé par des dosages veineux thyroïdiens sériques quantitatifs. Dans les cas de résultats intermédiaires, par exemple un taux de T4 bas mais un taux de TSH inférieur au seuil, un second test de dépistage par piqûre au talon est recommandé.
Les valeurs seuils pour la TSH ont changé au fil des ans, passant de 20 à 40 mU/l lors de l’introduction du dépistage néonatal à 7 à 10 mU/l au cours des dernières années.36 De manière prévisible, une augmentation de l’incidence d’HC primaire apparaît lorsque les seuils de TSH sont abaissés, avec une détection accrue des formes légères. En effet, avant l’ère du dépistage, l’incidence de l’HC était estimée à 1 : 6700 naissances37; l’utilisation de tests plus sensibles et des différentes stratégies de dépistage néonatal a permis la détection précoce d’un plus grand nombre de cas d’HC. Actuellement, cette incidence est estimée à environ 1 : 3000 à 4000 naissances.38
Conclusion
En Algérie, nous ne disposons pas à l’échelle nationale de données épidémiologiques en vue de déterminer la nécessité d’un dépistage néonatal systématique de cette pathologie. Nous supposons, tout de même, une fréquence relativement élevée de l’HC dans notre pays qui serait probablement liée, d’une part à l’existence d’une endémie goitreuse prédisposante par carence iodée maternelle, et d’autre part à la fréquence de la consanguinité dans notre contexte social. Enrayer des handicaps évitables justifie alors l’intérêt majeur d’un projet d’instauration du dépistage de cette affection chez tous les nouveau-nés. En attendant, nos médecins doivent rester vigilants, détecter les signes cliniques de l’hypothyroïdie congénitale et demander un dosage de la TSH au moindre doute.
Références
1- Alexander EK et al. Timing and magnitude of increases in Levothyroxine
requirements during pregnancy in women with hypothyroidism. N Eng J Med. 2004;351(3):25–33.
2- Williams GR. Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol. 2008;20(6):784–94.
3- Zoeller RT, Rovet J. Timing of thyroid hormone action in the developing brain: clinical observations and experimental findings. J Neuroendocrinol. 2004;16:809–18.
4- Fisher DA. The importance of early management in optimizing IQ in infants with congenital hypothyroidism. J Pediatr 2000 ; 136 : 273-4.
5- Beck-Peccoz P, Rodari G, Giavoli C, Lania A. Central hypothyroidisma neglected thyroid disorder. Nat Rev Endocrinol. 2017;13(10):588–98
6- Szinnai G. Clinical genetics of congenital hypothyroidism. Endocr Dev. 2014a;26:60–78.
7- Devos H et al. A search for the possible molecular mechanisms of thyroid dysgenesis: sex ratios and associated malformations. J Clin Endocrinol Metab. 1999;84(7):2502–6.
8- Knobel M, Medeiros-Neto G. An outline of inherited disorders of the thyroid hormone generating system. Thyroid. 2003;13(8):771–801.
9- Ordookhani A et al. High prevalence of consanguineous and severe congenital hypothyroidism in an Iranian population. J Pediatr Endocrinol Metab. 2004;17(9):1201–9.
10- Olivieri A et al. A population-based study on the frequency of additional congenital malformations in infants with congenital hypothyroidism: data from the Italian Registry for Congenital Hypothyroidism (1991-1998). J Clin Endocrinol Metab. 2002;87(2):557–62.
11- De Felice M, Di Lauro R. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev. 2004;25(5):722–46.
12- Perry R et al. Discordance of monozygotic twins for thyroid dysgenesis: implications for screening and for molecular pathophysiology. J Clin Endocrinol Metab. 2002;87(9):4072–7.
13- Castanet M et al. Linkage and mutational analysis of familial thyroid dysgenesis demonstrate genetic heterogeneity implicating novel genes. Eur J Hum Genet. 2005;13(2): 232–9.
14- Amendola E et al. A mouse model demonstrates a multigenic origin of congenital hypothyroidism. Endocrinology. 2005;146 (12):5038–47.
15- Vilain C et al. Autosomal dominant transmission of congenital thyroid hypoplasia due to lossoffunction mutation of PAX8. J Clin Endocrinol Metab 2001; 86:234.
16- Doyle DA, Gonzalez I, Thomas B, Scavina M. Autosomal dominant transmission of congenital hypothyroidism, neonatal respiratory distress, and ataxia caused by a mutation of NKX21. J Pediatr 2004; 145:190.
17- Castanet M, Park SM, Smith A, et al. A novel lossoffunction mutation in TTF2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet 2002; 11:2051.
18- LadoAbeal Jet al. A family with congenital hypothyroidism caused by a combination of lossoffunction mutations inthe thyrotropin receptor and adenylate cyclasestimulating G alphaprotein subunit genes. Thyroid 2011; 21.
19- Senée V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet 2006; 38:682.
20- De Filippis T et al . JAG1 LossOfFunction Variations as a Novel Predisposing Event in the Pathogenesis of Congenital Thyroid Defects. J Clin Endocrinol Metab 2016; 101:861.
21- Carré A et al. Mutations in BOREALIN cause thyroid dysgenesis. Hum Mol Genet 2017; 26:599.
22-Targovnik HM, Citterio CE, Rivolta CM. Iodide handling disorders (NIS, TPO, TG, IYD). Best Pract Res Clin Endocrinol Metab. 2017;31(2):195–212.
23- Dumitrescu AM et al. Impaired sensitivity to thyroid hormone: defects of transport, metabolism and action. Endotext [Internet]. South Dartmouth: MDText.com, Inc.; 2015 Aug 20.
24- GaudinoR et al.Proportion of varioustypes of thyroid disorders among newborns with congenital hypothyroidism and normally located gland: a regional cohort study. Clin Endocrinol. 2005;62(4):444–8.
25- Connelly KJ et al. Congenital hypothyroidism caused by excess prenatal maternal iodine ingestion. J Pediatr. 2012;161(4):760–2.
26- Atwell TDet al. Neonatal thyroid function after administration of IV iodinated contrast agent to 21 pregnant patients. AJR Am J Roentgenol 2008; 191:268
27- Zakarija M, McKenzie JM, Eidson MS. Transient neonatal hypothyroidism: characterization of maternal antibodies to the thyrotropin receptor. J Clin Endocrinol Metab 1990; 70:1239.
28- Huang SAet al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med 2000; 343:185.
29- Muzza M, Fugazzola L. Disorders of H2O2 generation. Best Pract Res Clin Endocrinol Metab. 2017;31(2):225–40.
30- Castanet M et al. Natural history and management of congenital hypothyroidism with in situ thyroid gland. Horm Res Paediatr. 2015;83(2):102–10.
31- LaFranchi SHet al. Neonatal hypothyroidism detected by the Northwest Regional Screening Program. Pediatrics 1979; 63:180.
32- Dussault JH, Coulombe P, Laberge C, et al. Preliminary report on a mass screening program for neonatal hypothyroidism. J Pediatr 1975; 86:670.
33- Mitchell ML, Larson CA. Neonatal screening for thyroid disease. In: Braverman LE, editor. Diseases of the thyroid. Humana Press, Totowa; 2003. p. 53–62.
34- Asami Tet al. Congenital hypothyroidism with delayed rise in serum TSH missed on newborn screening. Acta Paediatr Jpn 1995; 37:634
35- van Tijn DA, et al. Neonatal detection of congenital hypothyroidism of central origin. J Clin Endocrinol Metab 2005; 90:3350.
36- Lain S et al. Are lower TSH cutoffs in neonatal screening for congenital hypothyroidism warranted? Eur J Endocrinol. 2017;177(5):D1–D12.
37- Grosse SD, Van Vliet G. Prevention of intellectual disability through screening for congenital hypothyroidism: how much and at what level? Arch Dis Child. 2011;96:374–9.
38- Ford G, LaFranchi SH. Screening for congenital hypothyroidism: a worldwide view of strategies. Best Pract Res Clin Endocrinol Metab 2014; 28:175.