La Maladie de Gaucher – Vue d’Ensemble
Yanis AFIR
La maladie de Gaucher est une maladie lysosomale rare caractérisée par un déficit en glucocérébrosidase, qui entraîne l’accumulation de glucocérébrosides au sein des lysosomes macrophagiques ayant pour conséquence des dysfonctions multiples par des mécanismes complexes. Cliniquement, elle se caractérise essentiellement par une hépato-splénomégalie, une atteinte osseuse et, pour certaines formes, une atteinte neurologique. C’est l’un des rares déficits métaboliques héréditaires pour lequel il existe un traitement substitutif. Dans la majorité des cas, le pronostic vital n’est pas engagé mais le pronostic fonctionnel peut être sérieusement affecté.
Le présent article aspire modestement de faire un bref tour d’horizon de la maladie pour mettre en exergue ses aspects les plus importants.
Introduction
La maladie de Gaucher fait partie des maladies lysosomales. Pour rappel, le lysosome est un organite formé à partir du réticulum endoplasmique, contenant de nombreuses enzymes, appelées hydrolases acides, qui digèrent les macromolécules (glucides, protéines, lipides, ADN, ARN, etc.) absorbées ou produites par la cellule.
Les maladies lysosomales, en anglais Lysosomal Storage Diseases, sont des affections dues au déficit héréditaire de la fonction d’une ou de plusieurs des enzymes lysosomales ou de leurs protéines activatrices et dont la conséquence est l’accumulation des macromolécules non dégradées au sein de différentes cellules. Ce sont des maladies multisystémiques et évolutives.5
Bien que rares individuellement, les maladies lysosomales ont une fréquence cumulée estimée à 1 cas pour 5000 naissances.5 Parmi elles, la maladie de Gaucher est la plus fréquente (15%).1
Définition et généralités
La maladie de Gaucher est une maladie lysosomale rare, due essentiellement à une mutation du gène GBA1 (chromosome 1q21), entraînant un déficit dans l’activité de la glucocérébrosidase (ou glucosylcéramidase ou β-glucosidase acide, ou simplement Gcase). Elle est de transmission autosomique récessive. Plus de 400 mutations différentes ont été décrites, dont 80% sont des substitution d’un seul nucléotide.2 Exceptionnellement, elle peut être due à un déficit de l’activateur de la GCase, la saposine C.
La maladie fut décrite pour la première fois par Philippe Charles Ernest Gaucher, en 1882, chez une jeune femme souffrant d’une hépato-splénomégalie massive sans leucémie.6 Vingt ans plus tard, Nathan Brill démontre le caractère autosomique récessif de la maladie et lui donne l’éponyme de « Maladie de Gaucher ». Ce n’est que dans les années 60 que Roscoe Brady découvre le mécanisme physiopathologique à l’origine de la maladie et la lie au déficit de la Gcase.7
L’incidence de la maladie est estimée à environ 1 cas pour 50,000 naissances dans la population générale. Elle est plus fréquente dans la population juive ashkénaze où elle peut atteindre 1 cas pour 800 naissances.1
Quatre-vingt-dix pourcent des patients ont une maladie de Gaucher de type 1, dite forme non-neurologique (les formes cliniques sont détaillées plus bas), nettement plus fréquente chez la population juive ashkénaze. La maladie de Gaucher de type 2 est dite neuronale aiguë et touche tous les groupes ethniques, avec une incidence estimée à 1 cas pour 150,000 naissances.2 Quant au type 3, dit neuronal chronique, il est également pan-ethnique mais particulièrement fréquent dans les populations européennes nordiques, en Egypte et en Asie de l’est.2 Son incidence est estimée à 1 cas pour 200,000 naissances, donc moins fréquent que le type 2, mais sa prévalence est nettement supérieure à celle de ce dernier en raison de la survie plus longue des patients.2
Physiopathologie
La maladie de Gaucher résulte le plus souvent d’un déficit en glucocérébrosidase (Gcase), enzyme lysosomale dont le principal substrat est le glucocérébroside (Gcer), un composant de la membrane cellulaire. Rarement, le déficit peut concerner la protéine saposine C (mutation du gène PSAP1) qui se charge de présenter le Gcer à la Gcase.2
En conséquence il y a accumulation du Gcer au sein du lysosome de certaines cellules, notamment les macrophages de type M2 qui jouent un rôle important dans la dégradation de la membrane des hématies et des leucocytes, particulièrement riches en glucosphingolipides.6
Le Gcer accumulé au sein de ces macrophages prend l’aspect d’agrégats fibrillaires torsadés et leur donne l’image caractéristique de cytoplasme dit en « papier froissé ». Ils sont alors appelés cellules de Gaucher1 (fig. 1).
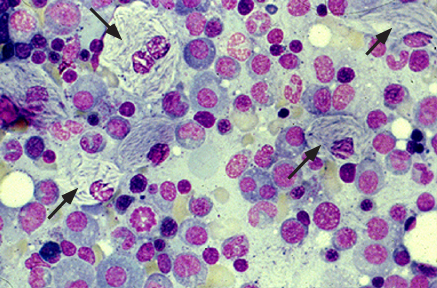
Figure 1 : Cellules de Gaucher vues au microscope optique sur ponction de moelle osseuse.
Image tirée de D. Hughes, E. Sidransky. Gaucher disease: Pathogenesis, clinical manifestations, and diagnosis (2019). S. Hahn (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
Pour des raisons encore inconnues, les cellules de Gaucher, ainsi que les macrophages environnant, se mettent à sécréter des enzymes lysosomales, des molécules inflammatoires telles que l’IL6, l’IL8, l’IL10 ou les MIPs (Macrophage Inflammatory Proteins), et des facteurs chémotactiques tels que le CXCL 2, 9, 10, 11 et le CCL18.
Les cellules de Gaucher infiltrent tout l’organisme. Elles sont particulièrement présentes au niveau de la rate, du foie et de l’os. Leur accumulation au niveau de la rate et du foie est responsable d’hépatomégalie et de splénomégalie. Cette dernière entraine la séquestration de cellules sanguines, avec pour conséquence des thrombopénies, voire des pancytopénies.
Au niveau osseux, l’infiltration massive des cellules induit une expansion de la cavité médullaire au détriment du tissu osseux9 ; ceci, associé à la dysfonction des ostéoblastes provoquée par les molécules inflammatoires, induit une diminution de la densité minérale et une fragilisation osseuse. De plus, la compression des vaisseaux qui résulte de l’infiltration prédispose aux infarctus osseux.8 Enfin, des fibroses de la moelle osseuse entravent l’hématopoïèse, participant ainsi également à la pathogénie des cytopénies.2
Les données nouvelles indiquent que les mécanismes pathogéniques de la maladie de Gaucher sont multiples et loin d’être tous élucidés. À titre d’exemple, en 2010, Mistry et al. ont identifié une voie métabolique alternative pour le Gcer qui est favorisée lorsque la voie classique de la Gcase est déficiente.17 Dans cette voie, le Gcer est transformé en glucosphingosine par une céramidase. Au sein du cytoplasme, la glucosphingosine est à son tour métabolisée par une autre Gcase, active à pH neutre (gène GBA2), en sphingosine et sphingosine-1-phosphate (S1P).6
La sphingosine serait particulièrement toxique pour l’os et les neurones. C’est d’ailleurs la principale hypothèse expliquant l’atteinte neurologique dans la maladie puisque les cellules de Gaucher ne pénètrent pas dans le cerveau.6 En effet, les études in vitro ont démontré que la sphingosine augmentait la libération de Ca2+ intracellulaire induisant une apoptose des neurones.5, 13
A noter que le turnover du Gcer dans les neurones est très faible et son accumulation n’est significative que lorsque l’activité de l’enzyme est très basse, c’est-à-dire uniquement pour certains types de mutations (voir plus bas).
La S1P a également un rôle dans la différenciation et la migration de plusieurs cellules comme les lymphocytes et les macrophages, contribuant sans doute à l’état d’inflammation observé.6
De même, le déséquilibre du ratio sphingosine/Gcer est à l’origine d’une perturbation de la formation de la barrière épidermique, responsable de l’ichtyose congénitale observée dans certains phénotypes de la maladie (voir plus bas).
Maladie de Gaucher et syndrome parkinsonien
Une prédisposition des patients atteints de la maladie de Gaucher pour le syndrome parkinsonien a été observée. Elle est probablement due au fait que la dysfonction de l’enzyme Gcase perturbe la dégradation lysosomale d’une protéine nommée α-synucléine, induisant l’accumulation de ses oligomères. Ceux-ci sont ensuite stabilisés par les Gcer et s’accumulent pour former des corps de Lewy. À noter que l’α-synucléine inhibe à son tour la Gcase, induisant ainsi un cercle vicieux.
Aussi, une seule mutation du gène GBA suffit à prédisposer au syndrome parkinsonien, même si 2 mutations sont nécessaires pour l’apparition de la maladie de Gaucher.
Génétique
Comme mentionné plus haut, la maladie de Gaucher est à transmission autosomique récessive et est essentiellement liée à une mutation du gène GBA1 sur le chromosome 1 (1q21). Parmi les nombreuses mutations décrites, 3 sont particulièrement fréquentes :
La mutation N370S (Asn370 → Ser) est la plus fréquente, particulièrement retrouvée dans la population juive ashkénaze. La protéine Gcase s’en trouve altérée mais garde une activité résiduelle, expliquant sans doute que la majorité des patients présentant cette mutation a une maladie de Gaucher de type 1, sans atteinte neurologique. En d’autres termes, la présence de cette mutation dans au moins un des allèles suffit à procurer une activité enzymatique suffisante pour protéger des effets neurologiques, et ce, quel que soit le type de mutation que peut subir l’autre allèle.7 Aussi, les formes homozygotes de N370S sont celles avec les manifestations les moins sévères.9, 12
La mutation L444P (Leu444 → Pro) est particulièrement fréquente dans les populations nordiques européennes. Le phénotype a tendance à être plus sévère, souvent de type 3, et particulièrement en cas de forme homozygote.
Enfin, la mutation c.84insG correspond à l’insertion d’une seconde Guanine au niveau du 84ème nucléotide. C’est une mutation non-sens où aucune protéine n’est synthétisée. Les patients atteints sont hétérozygotes, car la forme homozygote serait létale in utero.
Clinique
La maladie de Gaucher comprend une hépato-splénomégalie ainsi qu’une atteinte osseuse et médullaire chez quasiment tous les patients. L’atteinte neurologique s’y associe dans certaines formes. La sévérité de la maladie est très variable, pouvant être totalement asymptomatique ou rapidement létale. Le début de la symptomatologie peut survenir à n’importe quel âge. De manière générale, plus le déficit enzymatique est profond, plus la maladie est sévère et survient précocement.5
Il est également utile de mentionner que les différentes atteintes n’évoluent pas au même rythme, les atteintes hématologiques et viscérales ayant généralement l’évolution la plus rapide.15
Au final, il est classique de distinguer 3 types dans la maladie de Gaucher, le type 1 étant la forme non-neurologique, le type 2, le plus sévère, comportant des manifestations neurologiques aiguës et sévères, et le type 3 des manifestations neurologiques chroniques et moins sévères. Cependant, il est nécessaire de garder à l’esprit qu’il existe un continuum de phénotypes qui se superposent partiellement.
Maladie de Gaucher de type 1
Classiquement, le type 1 est défini par l’absence d’atteinte neurologique. C’est le type le plus fréquent (90% des cas) ; l’âge moyen du diagnostic est entre 10 et 20 ans6 mais peut se manifester dès 1 an, ou rester asymptomatique tout au long de la vie.2 Il peut parfois mettre en jeu le pronostic fonctionnel, mais exceptionnellement le pronostic vital.
De manière générale, la maladie progresse rapidement chez l’enfant et plus lentement chez l’adulte. De ce fait, toute altération rapide chez un adulte doit faire rechercher une cause concomitante ou faire évoquer un diagnostic différentiel. Aussi, une stabilisation de la maladie et même une régression spontanée ont été rapportées chez des adultes non traités.3
La symptomatologie se résume essentiellement en une hépato-splénomégalie et une atteinte osseuse et médullaire avec leurs conséquences respectives. Aussi, on note une asthénie dans la moitié des cas ainsi qu’un retard de croissance ou un retard pubertaire chez environ le tiers des enfants.
La splénomégalie, retrouvée chez 90% des patients, est due à l’accumulation de macrophages au sein de la rate. Sa taille peut être très importante, atteignant plusieurs dizaines de fois le volume splénique normal.2, 9 Elle peut être alors symptomatique ou se compliquer d’infarctus splénique, rare, ou de rupture de la rate, exceptionnelle.
L’hépatomégalie est notée chez 70% des patients, elle est également due à l’accumulation de macrophages au niveau du foie. Elle est généralement moins importante que la splénomégalie, le foie ayant un volume 2 à 3 fois plus important que son volume normal.2 L’évolution vers la cirrhose est rare.1 Une lithiase cholestérolique est souvent retrouvée.10
Il est important de noter que le diagnostic de la maladie de Gaucher devant une hépato-splénomégalie n’est pas évident et est souvent retardé, car d’autres pistes sont privilégiées. Il faut savoir suspecter le diagnostic en l’absence d’étiologie évidente et surtout si l’hépato-splénomégalie est accompagnée de thrombopénie, d’hyper-γ-globulinémie ou encore d’hyperferritinémie.
Aussi, chez plus de 40% des patients, l’hépato-splénomégalie se présente sous la forme de tuméfactions focales sous forme de nodules, appelés Gaucheromes. Là encore, il faut éliminer en priorité les diagnostics différentiels, notamment tumoraux.6, 9
L’atteinte hématologique est fréquente. Il s’agit le plus souvent d’une thrombopénie, responsable d’un syndrome hémorragique, à laquelle sont associés des troubles de la coagulation ou de l’hémostase primaire et une thrombopathie.
Les autres lignées peuvent être également atteintes ; une anémie est retrouvée chez 30 à 50% des patients, la leucopénie est plus rare. Notons que dans ce dernier cas il s’agit plus souvent d’une lymphopénie que d’une neutropénie, orientant vers le diagnostic de maladie de Gaucher plutôt que vers une hémopathie maligne.2
L’examen biologique montre souvent une hyper-γ-globulinémie polyclonale, et parfois un pic monoclonal. Aussi, des auto-anticorps (antinucléaires, antiphospholipides) ont été décrits, habituellement sans traduction clinique.1
L’atteinte osseuse est la principale source de morbidité et celle qui conditionne le pronostic fonctionnel.
Les premières manifestations, avant l’apparition des signes cliniques, sont représentées par une infiltration osseuse et des troubles du remodelage osseux avec élargissement de la région métaphyso-diaphysaire de l’extrémité distale du fémur, donnant un aspect en « fiole d’Erlenmeyer »8 (Fig. 2). Même si cet aspect n’est pas pathognomonique de la maladie de Gaucher, les diagnostics différentiels sont limités.2
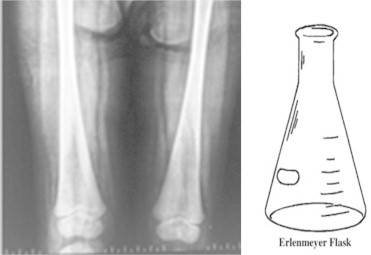
Figure 2 : Déformation osseuse en « fiole d’Erlenmeyer »
Image tirée de Nagral, A. (2014). Gaucher Disease. Journal Of Clinical And Experimental Hepatology, 4(1), 37-50.
L’atteinte osseuse est ponctuée de crises douloureuses aiguës et intenses correspondant à des infarctus osseux. Elles sont plus fréquentes chez l’enfant où elles peuvent mimer une ostéomyélite.6
Aussi, il est fréquent d’observer une ostéonécrose aseptique de la tête fémorale, une ostéoporose ainsi que des fractures pathologiques. Ces évènements peuvent être très invalidants pour le patient. L’atteinte osseuse est la plus lente à se normaliser après instauration de l’enzymothérapie14 et les lésions une fois acquises sont généralement irréversibles.8
Autres atteintes : à côté des manifestations classiques de la maladie, une multitude d’autres organes peuvent être impliqués. Nous citons à titre indicatif, et de manière non exhaustive, l’atteinte pulmonaire interstitielle, qui peut se voir lorsque les cellules de Gaucher infiltrent l’interstitium, où elles peuvent occlure les capillaires pulmonaires et ainsi engendrer une hypertension portale, et se compliquant rarement d’un syndrome hépato-pulmonaire. Aussi, une atteinte rénale, traduite par une hématurie et une protéinurie, peut rarement se voir lorsque les cellules de Gaucher infiltrent les glomérules. La peau peut être touchée, avec une hyperpigmentation aux tibias et aux joues.
à côté des manifestations classiques de la maladie, une multitude d’autres organes peuvent être impliqués. Nous citons à titre indicatif, et de manière non exhaustive, l’atteinte pulmonaire interstitielle, qui peut se voir lorsque les cellules de Gaucher infiltrent l’interstitium, où elles peuvent occlure les capillaires pulmonaires et ainsi engendrer une hypertension portale, et se compliquant rarement d’un syndrome hépato-pulmonaire. Aussi, une atteinte rénale, traduite par une hématurie et une protéinurie, peut rarement se voir lorsque les cellules de Gaucher infiltrent les glomérules. La peau peut être touchée, avec une hyperpigmentation aux tibias et aux joues.
Comme mentionné précédemment, un retard statural ou pubertaire est fréquent chez les enfants. Cependant celui-ci reste le plus souvent modéré, dans le cas contraire il faut éliminer une autre cause.
Un risque accru de cancers, particulièrement hématologiques (leucémies, lymphomes et myélome multiple) a été rapporté.
Les anomalies métaboliques ont été décrites telles qu’une résistance à l’insuline, des troubles du métabolisme lipidique ou une augmentation du métabolisme de base.
Enfin, même s’il est classique de dire que le type 1 ne s’accompagne pas d’atteinte neurologique, il a été constaté une prévalence des neuropathies périphériques supérieure à celle de la population générale, et comme expliqué plus haut, les patients atteints de la maladie de Gaucher ont un risque accru de développer un syndrome parkinsonien (× 4 à 20). Celui-ci est généralement précoce, agressif et résistant aux agonistes dopaminergiques.1 Cependant, la plupart des patients ne développent pas de syndrome Parkinsonien, suggérant que la maladie ne ferait qu’accroitre le risque chez des personnes déjà prédisposées.2
Maladie de Gaucher de type 2
C’est la forme neuronopathique aiguë, elle représente 1% des cas.9 Elle se traduit par une atteinte neurologique précoce et sévère, débutant la première année, associée à une atteinte systémique.
L’atteinte neurologique comprend une triade très évocatrice associant1 : une paralysie oculomotrice avec strabisme, typiquement la première manifestation,2 et plus tard des opisthotonos et des signes bulbaires (en particulier des troubles sévères de la déglutition).
A un stade plus évolué apparaissent divers signes tels qu’une hypertonie (avec trismus) et une rigidité, des spasmes laryngés responsables d’apnées, et des convulsions. Le développement psychomoteur est également altéré.
Les manifestations viscérales sont similaires au type 1 hormis l’atteinte osseuse qui est absente dans le type 2.
Le décès survient généralement avant l’âge de 2 ans, des suites d’une inhalation massive ou d’une apnée prolongée. Le traitement enzymatique est inefficace.
Une forme particulière de la maladie est la forme fœtale, la plus rare et la plus grave. Elle se révèle souvent par la mort in utero due à une anasarque fœto-placentaire. L’enfant peut toutefois survivre après la naissance et présenter alors une ichtyose congénitale (bébé collodion).
Maladie de Gaucher de type 3
C’est la forme neuronopathique subaiguë ou chronique. En plus de l’atteinte viscérale similaire à celle du type 1, on observe des manifestations neurologiques se résumant essentiellement en une paralysie oculomotrice supra-nucléaire, qui peut rester longtemps le seul symptôme ou s’associer à des myoclonies, une démence ou même une ataxie cérébelleuse. Son absence n’élimine pas le diagnostic.
La symptomatologie apparaît généralement avant 20 ans, plus tard que pour le type 2 mais parfois au même âge, rendant le diagnostic délicat. Aussi, les signes neurologiques peuvent apparaître longtemps après l’atteinte viscérale et donc souvent chez des patients initialement étiquetés comme de type 1.
On peut diviser le type 3 en trois formes, même si cette approche est loin d’être consensuelle tant les manifestations se ressemblent et se chevauchent2 : le type 3a est caractérisé par une atteinte neurologique sévère et des manifestations viscérales légères ; à l’inverse, dans le type 3b les manifestations neurologiques sont limitées, mais la viscéromégalie est massive et l’atteinte osseuse sévère, entraînant une cyphose. Celle-ci n’est pas due à la destruction osseuse et pourrait être liée à l’effet mécanique de la splénomégalie, à un effet neurologique ou d’origine génétique par mutation d’un gène voisin du gène GBA.15
Le type 3c quant à lui est rare et unique. Il associe une paralysie oculomotrice et une atteinte cardiovasculaire se manifestant par des calcifications aortiques et valvulaires. Il est associé à la mutation D409H homozygote, retrouvée dans les populations du bassin méditerranéen et au Japon.12 On peut également retrouver des cardiomyopathies dues à l’infiltration du myocarde par les cellules de Gaucher.9
Les exceptionnels déficits en saposine C comportent quasiment toujours une atteinte neurologique comparable à celle du type 3.
Biomarqueurs
Les principaux biomarqueurs de la maladie de Gaucher sont la chitotriosidase et le CCL18.
La chitotriosidase, une enzyme dont la fonction est inconnue, est produite par les cellules de Gaucher. L’analyse de sa cinétique évolutive renseigne sur la réponse au traitement et aurait une valeur pronostique. Cependant l’activité chitotiosidase est soumise à une grande variabilité individuelle rendant l’interprétation difficile.
La CCL18 quant à elle est une chimiokine produite par différentes cellules immunitaires, dont les cellules de Gaucher. Cependant, des taux élevés peuvent également se voir au cours des maladies inflammatoires chroniques. Sa variabilité interindividuelle est moins importante que pour la chitotriosidase car il n’existe pas de polymorphisme génétique et son évaluation est indispensable lorsque le patient est déficitaire en chitotriosidase.
A côté de cela, il est utile de doser la ferritine, bien que non spécifique, car son taux pourrait être prédictif de la survenue de complications osseuses.
A noter que la sphingosine décrite précédemment pourrait servir de biomarqueur nettement plus sensible et plus spécifique.1
Diagnostic de certitude
La mise en évidence du déficit enzymatique est le seul test de certitude. Il repose sur la mesure de l’activité de la Gcase sur prélèvement sanguin (tube EDTA).
Dans le type 1, on observe généralement une activité enzymatique résiduelle (10 à 15% de la valeur normale). Les types 2 et 3 ont une activité enzymatique nettement inférieure mais ne peuvent être différenciés l’un de l’autre.
Un diagnostic prénatal est possible par mesure de l’activité enzymatique dans les villosités choriales mais n’est pas de pratique courante.
Un exceptionnel déficit en saposine C doit être recherché en cas d’activité GCase normale alors que le tableau clinique et les biomarqueurs sont en faveur du diagnostic, sa recherche se fait par séquençage du gène PSAP.
Le myélogramme n’est pas nécessaire pour confirmer le diagnostic de maladie de Gaucher mais il est souvent réalisé dans l’investigation initiale pour éliminer les diagnostics différentiels. Il montre des cellules de Gaucher très évocatrices mais difficiles à repérer pour un cytologiste non expérimenté, elles sont encore plus difficilement différenciées des cellules pseudo-Gaucher retrouvées dans certaines hémopathies malignes ou infectieuses (myélome multiple ou maladie de Waldenström, lymphomes ou leucémie myéloïde chronique, mycobactérioses atypiques).
Enfin, la recherche des mutations du gène GBA1 par biologie moléculaire apporte des informations utiles pour le pronostic, en raison des possibles corrélations génotype-phénotype.
Traitement
Les principaux objectifs du traitement sont l’amélioration des symptômes et la qualité de vie ainsi que la prévention des complications irréversibles. À cela s’ajoute l’optimisation de la croissance staturale chez l’enfant.4 La découverte des traitements spécifiques a révolutionné le pronostic de la maladie. Cependant, le traitement n’est pas justifié chez tous les patients, même si tous nécessitent un suivi régulier.
Les 2 principales armes thérapeutiques sont le traitement enzymatique substitutif et le traitement par réduction de substrat.
Le traitement de première intention est l’enzymothérapie substitutive. Plusieurs molécules ont été développées, globalement d’efficacité similaire1 et toutes avec un coût exorbitant.
Dans le type 1, le traitement est indiqué dans les formes sévères, c’est-à-dire chez les patients ayant une thrombopénie ou une anémie symptomatiques ou sévères, une viscéromégalie symptomatique ou une atteinte osseuse ou des autres organes. Il est également indiqué chez l’enfant en cas de retard de croissance ou de retard pubertaire.
Les patients répondent généralement bien au traitement, les premières améliorations étant constatées dès 6 mois et pouvant se poursuivre jusqu’à 10 ans. Hormis les nécroses et les remodelages osseux, la plupart des atteintes sont réversibles.
Le traitement est généralement bien toléré. Les réactions allergiques sont peu fréquentes. Certains patients développent des anticorps contre l’enzyme substitutive, habituellement sans traduction clinique ; leur recherche systématique n’est pas recommandée.4
Concernant le type 3, le traitement est indiqué dans tous les cas, y compris dans les formes asymptomatiques ayant un génotype prédisposant au type 3.1 Les atteintes viscérales et squelettiques répondent bien au traitement mais pas les troubles neurologiques car l’enzyme recombinante ne traverse pas la barrière hémato-encéphalique.
Enfin, pour le type 2, le traitement n’est généralement pas initié car inefficace de toute façon.
Le traitement par réduction de substrat est le traitement de deuxième intention dans la maladie de Gaucher. Il vise à diminuer la production des Gcer. Il est efficace sur la viscéromégalie mais moins sur les troubles hématologiques et osseux. De même, son efficacité sur les troubles neurologiques n’est pas démontrée, malgré son passage à travers la barrière hémato-encéphalique. À noter qu’il est formellement contre-indiqué en cas de grossesse.
Les indications précises et les conduites thérapeutiques, délibérément non détaillées dans le présent article, sont largement commentées dans les différentes recommandations internationales, que nous vous invitons à consulter.18-22
Nous finissons par une brève mention des autres armes thérapeutiques : la greffe de moelle osseuse pourrait théoriquement guérir la maladie mais à l’heure actuel, et étant donné l’efficacité du traitement substitutif et les risques de la greffe, ce traitement n’est réservé qu’à une minorité de patients. La splénectomie quant à elle, autrefois utilisée pour prévenir les cytopénies, est de moins en moins utilisée en raison de sa tendance à aggraver les signes osseux, pulmonaires et hépatiques de la maladie, en plus de ses risques propres.
Conclusion
En conclusion, il apparaît assez clairement que la maladie de Gaucher est autant embêtante à diagnostiquer que barbante à étudier (très judicieux de ne mentionner cela qu’à la fin de l’article !) ; et même si il y a peu de chance qu’elle soit rencontrée par un praticien général, le simple fait d’y penser et d’orienter correctement le patient peut lui éviter des complications fâcheuses, voire graves. Gardons la donc soigneusement dans un coin de notre esprit.
Références
1- Berger M, Stirnemann J. Maladie de Gaucher. EMC – Hématologie 2016;11(1):1-16 [Article 13-012-I-10].
2- Hughes D., Sidransky E. Gaucher disease: Pathogenesis, clinical manifestations, and diagnosis (2019). Hahn S. (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
3- Hughes D., Sidransky E. Gaucher disease: Initial assessment, monitoring, and clinical course (2019). Hahn S. (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
4- Hughes D., Sidransky E. Gaucher disease: Treatment (2019). Hahn S. (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
5- Atul B. Mehta. Common hereditary lysosomal storage diseases. BMJ Best Practice. Feb 2019.
6- Stirnemann et al. (2017). A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. International Journal of Molecular Sciences, 18(2), p.441.
7- Dandana, A. et al. (2015). Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology, 83(1), 13-23.
8- Mucci, J., & Rozenfeld, P. (2015). Pathogenesis of Bone Alterations in Gaucher Disease: The Role of Immune System. Journal Of Immunology Research, 2015, 1-6.
9- Huang WJ, Zhang X, Chen WW (2015). Gaucher disease: A lysosomal neurodegenerative disorder. Eur Rev Med Pharmacol Sci. 2015;19:1219–1226.
10- Nagral, A. (2014). Gaucher Disease. Journal Of Clinical And Experimental Hepatology, 4(1), 37-50.
11- Baris H.N., Cohen I.J., Mistry P.K. (2014) Gaucher Disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev. 2014;12(0 1):72–81.
12- Cassinerio, E., Graziadei, G., & Poggiali, E. (2014). Gaucher disease: A diagnostic challenge for internists. European Journal Of Internal Medicine, 25(2), 117-124.
13- Pastores, G. (2010). Neuropathic Gaucher disease. Wiener Medizinische Wochenschrift, 160(23-24), 605-608.
14- Charrow, J., & Scott, C. (2015). Long-term treatment outcomes in Gaucher disease. American Journal Of Hematology, 90, S19-S24.
15- Mistry, P. et al. (2015). Understanding the natural history of Gaucher disease. American Journal Of Hematology, 90, S6-S11.
16- Roshan Lal, T., & Sidransky, E. (2017). The Spectrum of Neurological Manifestations Associated with Gaucher Disease. Diseases, 5(1), 10.
17- Mistry et al. (2010). Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proceedings Of The National Academy Of Sciences, 107(45), 19473-19478.
18- Hughes, D. et al. (2007). Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. British Journal Of Haematology, 138(6), 676-686.
19- Weinreb, N. et al. (2004). Gaucher disease type 1: Revised recommendations on evaluations and monitoring for adult patients. Seminars In Hematology, 41, 15-22.
20- Eliglustat for treating type 1 Gaucher disease. National Institute for Health and Care Excellence. 2017.
21- Kaplan, P. et al. (2012). Revised recommendations for the management of Gaucher disease in children. European Journal Of Pediatrics, 172(4), 447-458.
22- Pastores GM, et al. (2004). Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 Suppl 5):4–14.