Iron Overload ‘All in Moderation, Nothing in Excess’
Hadya LAGGOUN
Iron is an essential trace mineral for many biological processes, including hemoglobin synthesis, oxidation-reduction reactions and cellular proliferation. Conversely, iron is extremely toxic when present in excessive amounts. Thereby, a tight regulation of iron balance is essential to avoid iron overload. This review highlights recent advances that have changed our understanding of iron metabolism and its regulation, in order to assimilate the pathophysiology of iron overload diseases and their consequences. An approach to evaluating individuals with suspected iron overload is presented here as well.
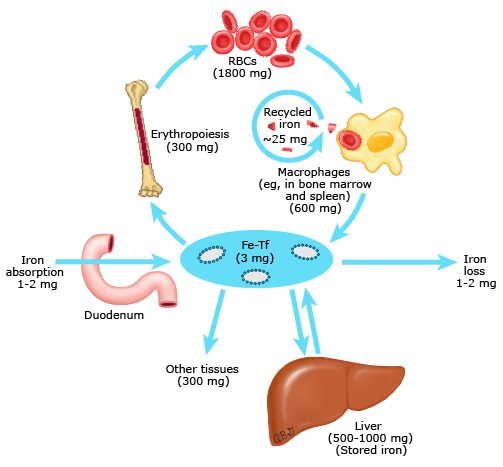
Body iron metabolism is a semi-closed system; its regulation involves the interaction of a number of specific proteins as well as the interplay between iron absorption, recycling and iron loss (figure 1).
There are no physiologic mechanisms to eliminate excess iron from the body1, 3, 4
Maintaining homeostatic balance requires only 1 to 2 mg of absorbed iron per day (i.e. one tenth of the iron content of the normal food bolus) to offset the equivalent losses that come out through the digestive, cutaneous and urinary routes. Because there are no physiologically regulated means of iron excretion, dietary iron absorption is highly regulated.
Firstly, iron absorption varies according to the form of iron in the diet. Indeed, heme dietary sources (fish, poultry, and meat) have a higher bioavailability than do non-heme (cereal, breads, fruits, and vegetables) ones (30 versus 10 percent). Thus, heme iron is absorbed more efficiently than non-heme iron; the molecular mechanisms of intestinal heme absorption remain poorly understood.5, 6
Moreover, intraluminal factors can affect absorption: ascorbic acid and meat sources enhance the absorption of non-animal sources of iron, whereas tannates (teas), bran foods rich in phosphates, and phytates inhibit it.
Additionally, non-heme iron is absorbed in the duodenum, where acidic gastric secretions enhance iron solubility. Ferrous iron (Fe2+) is taken at the mucosal side via a ferrous iron transporter, such as the Divalent Metal Transporter 1 (DMT1).7, 8 Conversely, luminal iron is prominently ferric (Fe3+) requiring reduction prior to uptake by the enterocyte, in part by the apical membrane reductase Duodenal CYTochrome B (DCYTB).9
Most of the iron taken up from either source is then stored in the form of ferritin and is lost upon sloughing of the senescent enterocyte. The iron that joins the circulation is transported through the basolateral membrane by the duodenal iron exporter, ferroportin. Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves ceruloplasmin, a known copper dependent multioxidase.
Notably, iron released from enterocytes (and macrophages, see below) binds to free sites on the specific plasmatic iron-transport protein: transferrin. Each transferring molecule can bind at most 2 iron atoms. This chelation renders the iron soluble and prevents iron-mediated free radical toxicity.
The distribution of iron to tissues relies almost entirely on the abundant plasma protein transferrin, but it is normally less than 45% saturated with iron.
Finally, the regulation of each previous step (reduction, absorption, storage, and transfer) is mediated by signals reflecting oxygen tension in enterocytes, intracellular iron levels, and systemic iron needs. The hypoxic environment of intestinal mucosa enhances iron absorption through its effects on the transcription factor Hypoxia Inducible Factor 2α (HIF-2α) which increases the expression of both key genes, DMT1 and DCYTB.10, 11 The enterocyte iron content also regulates iron absorption through its effects on Iron Regulatory Protein (IRP) types 1 and 2 and their subsequent effects on messenger RNAs (mRNAs) encoding DMT1, ferroportin, ferritin, and HIF-2α.12
Despite the cellular mechanisms noted above, intestinal iron absorption is primarily regulated at the enterocyte basolateral membrane through control of iron release into extracellular fluid and plasma. This systemic regulation is mediated by the hepatic peptide hormone hepcidin, which binds to the iron exporter ferroportin and induces its degradation; thus, decreasing the transfer of iron from enterocytes to the circulation.
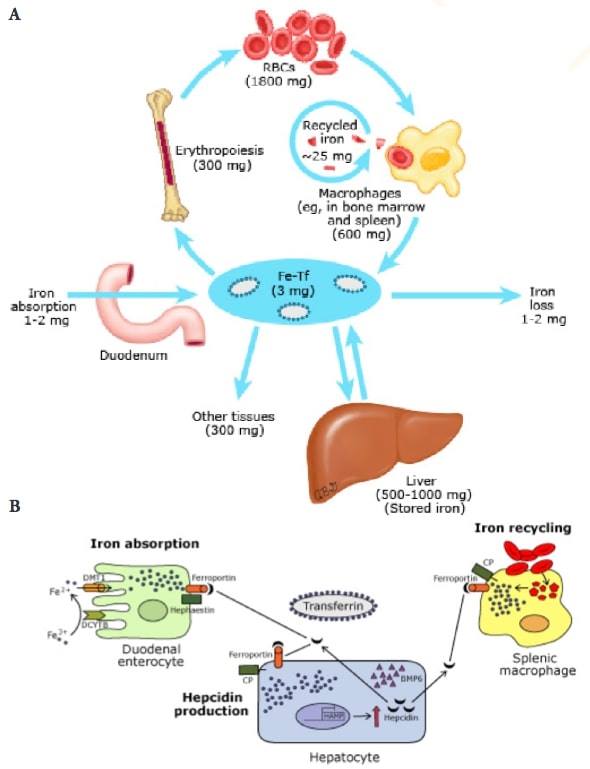
Figure 1: (A) Iron cycle. (B) Reduced iron absorption and recycling in iron overload
RBCs: red blood cells. Fe-Tf: transferrin-bound iron. DMT1: divalent metal transporter. DCYTB: duodenal cytochrome B. CP: ceruloplasmin. BMP6: bone morphogenetic protein 6. HAMP: hepcidin antimicrobial peptide (hepcidin gene).
Taken from Camaschella C. Regulation of iron balance. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc.
Iron economy is dominated by the production and turnover of red blood cells13
The normal iron content of the body is 3 to 4 grams, two thirds of which is in red blood cells (RBC) and is recycled from senescent RBCs’ breakdown by macrophages of the reticuloendothelial system.
In fact, blood iron circulates in 2 main forms: erythrocytic iron, which accounts for half of the body iron stores (about 2-2.5 g), and plasma iron, quantitatively much smaller than erythrocytic iron, around 3 to 7 mg in total.
Erythrocytic iron, a component of heme in hemoglobin, is derived from the bone marrow where the erythroid precursors express high levels of Transferrin Receptor 1 (TfR1). Erythropoietic activity is an important regulator of hepcidin expression (see “Hepcidin: the master regulator of systemic iron homeostasis”, below). On the other hand, plasma iron has a dual source : the absorption of dietary iron through the duodenum and, mostly (quantitatively), the plasma release of splenic iron originating from erythrophagocytosis.
In point of fact, unlike other body cells, reticuloendothelial cells obtain most of their iron from the phagocytosis of senescent erythrocytes. After release from heme, the resulting iron is released into the circulation through ferroportin and taken up by transferrin to regain the bone marrow. It can also be stored in ferritin according to the body needs and to the local concentration of hepcidin. Effectively, reticuloendothelial cells serve as the major hepcidin-regulated iron repository, as we are about to explain.
It is important to highlight that, at equilibrium, approximately 20 to 25 mg of iron are released daily from reticuloendothelial cells. Since the pool of circulating transferrin iron amounts to 3 mg, the rate of iron turnover by these cells is quite high (8 to 10 times). As a result, hepcidin-mediated changes in iron export can induce rapid and marked changes in serum iron concentrations.
Similar to reticuloendothelial cells, hepatocytes are an important site of iron storage in the form of ferritin. Most importantly, hepatocytes play a central role in iron homeostasis as the principal site of regulated production of the hormone hepcidin.
Hepcidin: the master regulator of systemic iron homeostasis1, 14, 15
The peptide hormone hepcidin functions as the “hypoferremia hormone”. In a remarkably streamlined regulatory mechanism, it orchestrates systemic iron flux by binding to the only known mammalian cellular iron exporter (ferroportin) on the surface of iron-releasing cells (macrophages and enterocytes), blocking cellular iron release into the circulation.
Hepcidin is feedback-regulated by iron status and strongly modulated by erythropoietic demand and signals reflecting oxygen tension or inflammation. Even so, the iron sensing and signaling pathway involving hepcidin is complex and not fully elucidated.
Transcription of hepcidin in response to increased plasmatic or tissue iron is mediated by Bone Morphogenetic Proteins (BMP) which requires hemojuvelin (HJV) as a coreceptor, and is SMAD-dependent. Supporting this model, BMP6 and/or Smad4 inactivation in mice causes severe iron overload with low hepcidin.16
Plasma iron-signal regulating hepcidin is provided by transferrin, which, upon binding iron, serves as a ligand for 2 hepatocellular receptors: TfR1 and TfR2. This signal appears to be modulated by the physical interaction of these 2 receptors with the hemochromatosis protein HFE: a major histocompatibility complex class I–like molecule without iron-transport properties. Whether and how the HFE-TFR2 complex, that is formed in the presence of increased diferric transferring, cooperates with the BMP-HJV-SMAD pathway for hepcidin activation is uncertain. However, in HFE-/- mice, the BMP pathway is indeed less active, and treatment with BMP6 appears to ameliorate iron overload.17
Erythropoietic activity has a greater influence on hepcidin expression than does body iron status. Since most iron is used by maturing erythroid cells, and to favor iron acquisition, hepcidin expression is markedly decreased in several conditions in which erythropoiesis is increased, including phlebotomy, hemolysis, erythropoietin administration, iron deficiency anemia, hypoxia, erythropoietic expansion and ineffective erythropoiesis. The existence of a regulator of iron absorption produced by erythroblasts was first proposed based on ferrokinetic studies. Candidate signaling molecules include Growth Differentiation Factor 15 (GDF15) and Erythroferrone (ERFE).18, 19
Under hypoxic conditions, hepcidin is downregulated to allow increased iron export through ferroportin.
Lastly, the inflammatory signal up-regulating hepcidin expression is largely mediated by IL-6, IL-1β and lipopolysaccharide.20 There is a crosstalk between the two pathways of hepcidin activation (inflammatory and iron-dependent) as demonstrated in the improvement of hepcidin control by compounds inhibiting BMP-SMAD pathways in inflammation.21
Iron overload is either the result of dysregulated iron-status signal or erythroid signal and the consequent inadequate hepcidin expression2, 13, 22
Knowledge regarding the complexities of human iron metabolism has increased significantly in the last two decades and the classification of iron overload diseases has become more detailed as shown in table 1.
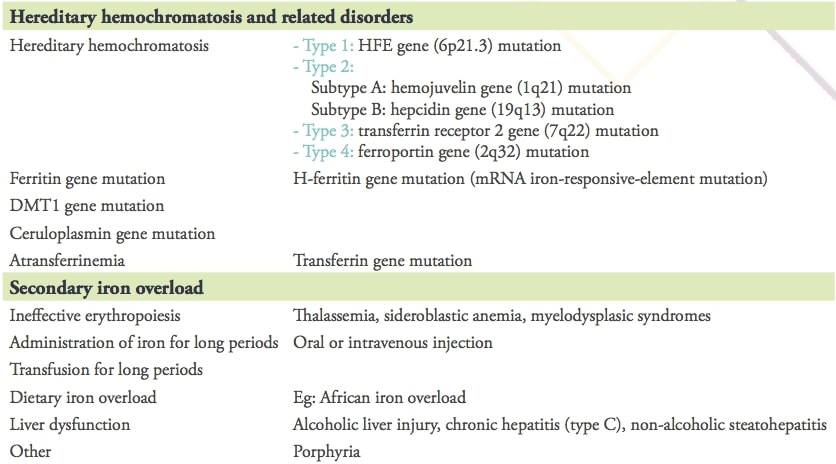
Table 1: Causes of iron overload
Reproduced from Y. Kohgo et al. Body iron metabolism and pathophysiology of iron overload. Int J Hematol. 2008; 88(1): 7–15.
Each iron-overload disorder by defect in the hepcidin–ferroportin axis represents mainly a form of primary iron overload and a subtype of hemochromatosis. By far, the most common cause in this category is type 1 hemochromatosis related to mutations of the HFE gene, typically homozygous C282Y/C282Y or compound heterozygous C282Y/H63D. Affecting caucasian populations, individuals with type 1 hemochromatosis can absorb as much as 2 to 4 mg of dietary iron per day (twice the normal rate).This increased absorption can result in an additional 3 mg per day in excess of needs. Over time, iron accumulation can thus occur in the range of 1g per year (10g per decade). This explains the typical presentation of adult males with signs and symptoms of hemochromatosis in the 4th to 5th decades, and the slightly later presentation of women, who have additional iron losses due to menstruation, pregnancy, and lactation.
Mutations in other iron regulatory genes have also been reported including hemojuvelin (HJV) gene, hepcidin gene (HAMP), ferroportin gene (SLC40A1), mutations in transferrin receptor 2 or ceruloplasmin. All these non-HFE gene hemochromatoses are rare or exceptional, however they are not limited to caucasian populations.
Despite the lack of genetic background, iron overload is commonly observed as a secondary condition. Frequently, it occurs in patients requiring long-term blood transfusion for anemia unrelated to iron deficiency. Examples include thalassemias (in which iron overload is compounded by increased absorption due to ineffective erythropoiesis), as well as sickle cell disease, other inherited anemias, aplastic anemia, myelodysplastic syndromes, other hematologic malignancies and hematopoietic cell transplantation.
In addition to these classical conditions, there are many diseases that show mild iron deposition or dysregulation of body iron distribution. Such conditions include liver diseases, especially alcoholic liver disease and chronic hepatitis.
Other less common causes of increased iron stores include gestational alloimmune liver disease (GALD), increased intake due to an iron-loaded diet such as in African iron overload and excessive use of iron supplements or therapeutic infusions of iron-containing products such as hemin, used to treat certain porphyrias.
Toxic effect of iron overload on organ function is due to the overproduction of ROS2, 23
As the body content of iron increases beyond that needed, the plasma iron-binding protein, transferrin, becomes saturated. Once transferrin saturation exceeds about 70%, non-transferrin-bound iron (NTBI) appears.
NTBI consists of iron-binding to other proteins and molecules, including albumin, citrate, acetate, and others. It is described as a high-speed iron species, with a parenchymal target which is unregulatable and potentially damaging. Indeed, this iron is taken up by cells that have active uptake mechanisms for NTBI; liver, heart, joints, skin, and endocrine organs appear to be especially susceptible. In these affected organs, NTBI is known to facilitate excessive Reactive Oxygen Species (ROS) generation exceeding the capacity of cellular antioxidant systems; which causes lipid peroxidation, oxidation of amino-acids with consequent protein–protein cross-links, protein fragmentation and DNA damage. Therefore, NTBI contributes to the development of tissue damage, inflammation and fibrosis.
Recently, the ion channel SLC39A14 (Solute Carrier Family 39 Member 14; ZIP14) was identified as the specific importer of NTBI in hepatocytes and pancreatic cells.24
Liver and heart demonstrate the highest propensity to accumulate iron. Thus, typical manifestations include, on one hand, hepatic involvement with biochemical abnormalities in liver function, inflammation, fibrosis and eventually cirrhosis; and on the other hand, cardiac involvement with cardiomyopathy, heart failure and/or arrhythmias.
Besides, clinical features include diabetes mellitus, arthropathy, hyperpigmentation (bronze skin) by pancreatic, joints and skin involvement respectively. Iron may also be deposited in other glands, such as the pituitary, gonadal, parathyroids and less commonly the thyroid, with consequent endocrine dysfunction (hypogonadism, impotence…).
By the time clinical findings have developed (hepatic fibrosis, heart failure, cardiac conduction defect), it is likely that significant iron deposition and tissue injury has occurred.
Importance of early diagnosis before symptomatic disease lies in preventing organ damage and limiting disease progression13, 25
The signs and symptoms of iron overload are insensitive and nonspecific. Therefore, early diagnosis of iron overload, before organ damage occurs, requires consideration of this possibility when the physician is faced with such common findings as chronic fatigue, joint pain, impotence, osteoporosis, diabetes and predisposition to infections.
The relative likelihood of iron overload versus other causes of organ dysfunction depends on a number of factors that affect the likelihood of possible diagnoses, including patient age, family history, other symptoms and other comorbidities. Thus, iron overload is more likely to be a cause of organ damage in middle aged men or postmenopausal women, those with a family history of hemochromatosis and those with a history of multiple RBC transfusions.
Iron overload may also be suspected in the presence of unexplained organ damage such as liver disease, cardiac disease, endocrine disease, or an incidental finding of increased serum ferritin or increased transferrin saturation (TSAT).
The diagnosis of iron overload requires clinical correlation of the sign and symptoms with an estimation of total body iron accumulation/stores2, 26, 27
In addition to a thorough clinical evaluation, all patients with suspected iron overload should have the following laboratory tests: a complete blood count (CBC) with RBC indices, routine iron studies and a metabolic panel including hepatic enzymes (alanine aminotransferase [ALT], aspartate aminotransferase [AST]).
Routine iron studies include serum iron, transferrin (also reported as Total Iron Binding Capacity [TIBC]), and ferritin; the transferrin saturation (TSAT) is calculated as the ratio of serum iron to TIBC and expressed as a percentage (TSAT = iron ÷ TIBC x 100).
The results most useful for evaluating iron overload are the ferritin and TSAT. A low or normal serum ferritin or TSAT is helpful in eliminating the possibility of iron overload. Ferritin levels above 200 ng per milliliter (449 pmol per liter) in women or 300 ng per milliliter (674 pmol per liter) in men without significant inflammation and transferrin saturation above 40% in women or 45% in men are consistent with iron overload but are not particularly specific.
There are several factors that may confound ferritin and TSAT measurements, including diet, acute infection or inflammation, malignancy and comorbidities, especially liver disease. It is important to distinguish these other causes of abnormally high ferritin/TSAT in order to avoid unnecessary invasive testing and/or delays in treatment. Consequently, it is ideal to obtain at least 2 independent measurements, and liver function testing is often helpful.
The CBC and iron studies are interpreted together, because the presence of anemia influences the evaluation. For instance, a normal CBC with increased ferritin and transferrin saturation is suggestive of hemochromatosis, microcytic anemia with increased ferritin and transferrin saturation is strongly suggestive of thalassemia whereas macrocytic anemia suggests that there is an underlying cause of anemia such as hemolysis or megaloblastic anemia.
Other testing such as hepatic or cardiac MRI or liver biopsy with iron staining are generally limited; on one hand to individuals with laboratory evidence of iron overload, in order to estimate the extent of iron overload and severity of tissue damage, which determines the need for and urgency of therapeutic interventions; or on the other hand, in those for whom there is diagnostic confusion, in order to definitively establish or exclude the presence of increased tissue iron.
Removal of iron with a course of therapeutic phlebotomy (at least 5 to 6 phlebotomies in individuals who do not have significant anemia or other comorbidities) with normalization of ferritin level is also in favor of an iron overload diagnosis.
Additional testing (eg, genetic testing for HFE mutations) is needed to determine the underlying cause(s) of iron overload so that it can be addressed with a long-term treatment plan.
The major treatments for iron overload include phlebotomy and chelation therapy
Means for both the early detection and treatment of iron overload are fortunately available now and, with appropriate therapy, many of the pathological effects of iron excess can be avoided. The mainstays of treating systemic iron overload are iron removal by phlebotomy in the absence of anemia (applicable in most forms of hereditary hemochromatosis) and chelation in the iron-loading anemias. Currently, research studies are ongoing to provide newer chelating agents and novel therapeutic options for iron overload. For example, hepcidin-like molecules have been shown to ameliorate iron overload in preclinical models of HH and β-thalassemia and are ongoing clinical evaluation.28
Over and above, it is known that there is abundant expression of DMT1 in the proximal tubule and collecting ducts of the kidney. This raises the possibility that there is an iron excretory pathway that is overcome by constitutive reabsorption. If this hypothesis is correct, pharmacologic blockade of this pathway could represent an important therapeutic maneuver for syndromes of iron overload.29
In conclusion, iron overload is an excess of systemic iron, leading to its progressive accumulation in vital organs. Patients with iron overload are often asymptomatic. When signs and symptoms occur, they are generally related to specific organ involvement. Fortunately, when iron overload is suspected after a history and physical examination, it can generally be diagnosed with low costs, non-invasive blood tests. The prognosis of patients with iron overload is extremely positive when diagnosed early and when treatment effectively reduces iron levels. Therefore, physicians should identify iron overload cases at an early stage and initiate prompt, suitable treatment.
References
1- Camaschella C. Regulation of iron balance. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
2- Bacon BR, Kwiatkowski JL. Approach to the patient with suspected iron overload. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
3- Coffey, R., & Ganz, T. Iron homeostasis: An anthropocentric perspective. Journal of Biological Chemistry 2017; 292(31), 12727–12734.
4- Muckenthaler, M. U. et al. A Red Carpet for Iron Metabolism. Cell 2017; 168(3), 344–361.
5- Shayeghi M et al. Identification of an intestinal heme transporter. Cell 2005; 122:789.
6- Fillebeen C et al. Mice are poor heme absorbers and do not require intestinal Hmox1 for dietary heme iron assimilation. Haematologica 2015 ; 100, e334– e337.
7- Illing AC et al. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J. Biol. Chem2012; 287: 30485–30496 5.
8- Wang CY et al.ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem 2012; 287: 34032–34043.
9- McKie AT et al. An iron-regulated ferric reductase associated with the absorption of dietary iron.Science 2001; 291(5509):1755-9.
10- Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab 2009; 9:152-64.
11- Mastrogiannaki M et al. HIF-2 alpha, but not HIF-1 alpha, promotes iron absorption in mice. J Clin Invest 2009;119:1159-66.
12- Galy B et al. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab 2008; 7:79-85.
13- Robert E. Fleming, and PremPonka. Mechanisms of Disease- Iron Overload in Human Disease.N Engl J Med 2012;366:348-59.
14- Camaschella C. Iron and hepcidin: a story of recycling and balance. Hematology Am SocHematolEduc Program 2013; 2013:1.
15- Ganz, T. Systemic iron homeostasis. PhysiolRev 2013; 93, 1721–1741.
16- Meynard D, Kautz L, Darnaud V, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 2009; 41:478.
17- Corradini E, Schmidt PJ, Meynard D, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 2010; 139:1721.
18- Tanno T, Noel P, Miller JL. Growth differentiation factor 15 in erythroid health and disease. CurrOpinHematol 2010; 17:184.
19- Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014; 46:678.
20- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037.
21- Theurl I, Schroll A, Sonnweber T, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 2011; 118:4977.
22- Yutaka Kohgo, Katsuya Ikuta, TakaakiOhtake,YoshihiroTorimoto, and Junji Kato. Body iron metabolism and pathophysiology of iron overload. Int J Hematol. 2008; 88(1): 7–15.
23- Brissot P, Ropert M, Le Lan C, Loreal O. Non-transferrin bound iron: a key role in iron overload and iron toxicity. BiochimBiophys Acta 2011 August 9.
24- Jenkitkasemwong, S., Wang, C. Y., Coffey, R., et al. SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab 2015. 22, 138–150.
25- Ginzburg, Y. Z., & Vinchi, F. Iron Overload. Transfusion Medicine and Hemostasis 2019; 433–436.
26- Wood JC. Guidelines for quantifying iron overload. Hematology Am SocHematolEduc Program 2014; 2014:210.
27- Jensen PD. Evaluation of iron overload. Br J Haematol 2004; 124:697.
28- Pietrangelo A. Hepcidin in human iron disorders: therapeutic implications. J Hepatol 2011;54:173-81.
29- Andrews NC. Forging a field: the golden age of iron biology. Blood 2008; 112:219.