Immune checkpoints inhibitors: A short review
Yanis AFIR
Since a decade now, the use of immune checkpoints inhibitors in clinical practice worldwide has truly revolutionized oncology and immunotherapy. Perhaps the best illustration of this impact is awarding the 2018 Medicine Nobel Prize to Allison and Honjo, two key actors of this long scientific journey. Beside being a quintessential part of our general medical culture, the topic concerns us even more as Algerian physicians or medical students given that the introduction of this therapy is expected in our country starting next year.
Here we shall take a glance at the main molecular actions of immune-checkpoints inhibitors, some clinical inquiries about their use, the resistance to the therapy and the main adverse effects.
Introduction
The idea of a relationship between cancer and the immune system is as old as oncology itself. In fact, the immune system was proposed as an “adjuvant” therapy long ago when William Coley observed a remarkable tumor burden regression in patients injected with Streptococcus pyogenes which was due to bacterial-induced inflammation (1).Since then, many studies tried to understand the processes governing the relationship between cancer cells and immunity effectors.
We know for sure that the immune system is appropriately equipped with many defensive weapons against tumor cells including T cells and various cytokines. However, tumors can block these defensive tools by co-opting certain immune pathways called “immune-checkpoints”.
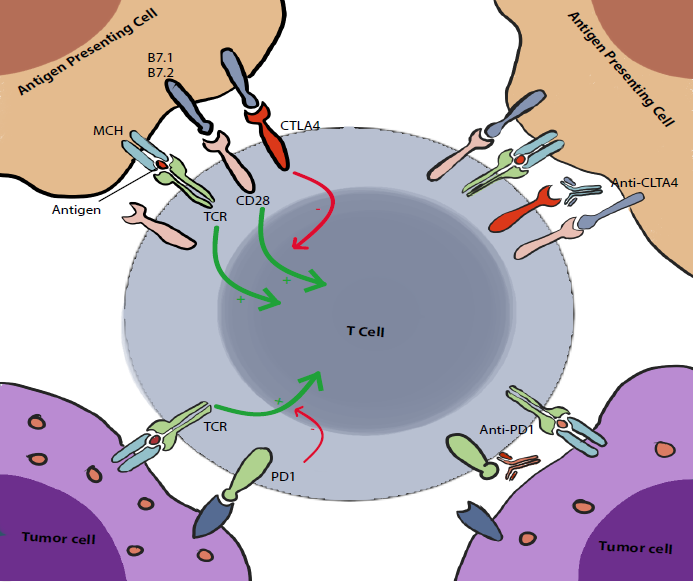
Immune checkpoints can be defined as “a plethora of inhibitory pathways hardwired into the immune system that are crucial for maintaining self-tolerance and modulating the duration and amplitude of physiological immune responses in peripheral tissues in order to minimize collateral tissue damage” (2).
To make it easy let’s remember that, in order to complete its task, LT4 cells need to be activated by various stimulatory signals. However, if left uncontrolled, these stimulatory signals may produce a reaction that is far greater than what is needed and could ultimately have deleterious effects by damaging the body cells more than the aggression itself and by making autoimmunity more likely.
To maintain equilibrium, the immune system also expresses inhibitory signals that modulate this excessive stimulation. These signals are known as checkpoints and are essentially, but not exclusively, represented by Cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and Programmed Cell Death Protein 1 (PD1).
Now let’s go back to cancer. Among the many mechanisms used by tumor cells to escape immunity, one of the most important is enhancing the activity of checkpoint-inhibitors. Thus, the equilibrium is shifted toward the
inhibition and immunologic cells can no longer fight cancer properly.
For this reason, and in order to help immunity win the fight, inhibiting the action of these checkpoints, thus unleashing the full potential of the immune system against the tumor cells was proposed.
This original approach relying on immune cells rather than targeting directly cancer was indeed a tremendous success as it demonstrated clinical activity in several malignancies, including melanoma, non-small cell lung
cancer, renal cell carcinoma, bladder cancer, head and neck squamous cell carcinoma, MSI (micro satellite instability) high colorectal carcinoma, Merkel cell carcinoma, and Hodgkin lymphoma (3).
CTLA4
The Cytotoxic T lymphocyte-associated antigen 4, also known as CD152 but commonly called CTLA4, was discovered in 1987 (4). It is expressed exclusively on T cells. Its main role is to regulate the amplitude of the early stages of T cell activation in the lymph nodes by counteracting the activity of the T cell co-stimulatory receptor, CD28 (2).
CD28 is a strong amplifier of TCR signaling on T cells, its ligands are CD80 (also known as B7.1) and CD86 (also known as B7.2). CTLA4 outcompetes CD28 by binding to these molecules with a higher affinity. This seems to be the main action of CTLA4, however we (I mean scientists :V) have reasons to believe that there are multiple other pathways and mechanisms of action including actively delivering inhibitory signals to the T cell and active removal of CD80 and CD86 from the cell surface. These are still under debate (2).
Anti-CTLA4 were the first antibodies synthesized for the aim of enhancing immune system response against tumor cells. Their use was first highly questioned due to a lack of tumor specificity targeting with this technique; plus, preclinical studies predicted high toxicity and adverse effects without any assurance of possible advantages.
However, by only partially blocking the effector and finding the proper therapeutic window, a team of researchers including J.P. Allison (one of the 2018 Nobel Prize winners) demonstrated for the first time the ability of CTLA4 antibodies to induce therapeutic antitumor immunity, that was in 1996 (5).
After that, two fully humanized CTLA4 antibodies, Ipilimumab and Tremelimumab, were synthesized. Between the two, Ipilimumab was the most studied molecule, mainly on patients with metastatic melanoma.
Since the beginning, clinical trials demonstrated that some poorly immunogenic tumors did not respond to anti- CTLA4 as a single agent. Interestingly, these tumors responded when anti-CTLA4 was combined with “cellular vaccines” (molecules used to enhance the immunogenicity of the cells) (6).
Thus, the clinical attitude was inclined to combine anti-CTLA4 with cellular vaccines. We had to wait until 2010 to have a major clinical trial demonstrating a significant benefit in long-term survival for patients with metastatic melanoma (7). The drug was approved as a treatment during the same year.
One feature of this treatment that contributed to labelling checkpoint-inhibition as revolutionary is the incredible durability of responses. Not only is overall survival enhanced, but so is long term survival (over 10 years sometimes) (8).
Another interesting fact about immune-checkpoint blocking is that the response is really slow and delayed (up to 6 months after initiating treatment); moreover, due to the increased immune cells infiltration, tumor size might increase in the beginning of treatment, before regressing later. These features contributed to the underestimation of the efficiency of these therapies before having long-term follow-up trials.
PD1
The Programmed Cell Death Protein 1, also known as CD 279 but commonly called PD1, was discovered in 1992 (by a team among which was Honjo, another 2018 Nobel Prize winner) (9). It was first noted that PD1 was expressed in dying cells (hence its name) but later, it was found that it has no role in apoptosis and is rather expressed in activated lymphocytes.
In contrast to CTLA4, PD1 acts mainly on already activated T cells. It plays a crucial role in limiting their activity on peripheral tissues (including tumoral tissues). Yet, similarly to CTLA4, it counters and modulates the positive stimulatory signals to T cells.
When PD1 binds to its ligands, PDL1 (PD1 ligand, also known as B7 H1 and CD274) and PDL2 (also known as B7 DC and CD273), it inhibits the cellular activating pathways mainly through the phosphatase SHP2. Other mechanisms are also involved including other phosphatases and modification of the T-cell/Target-cell contact duration (2).
It was also showed that there may be an interaction between PDL1 and CD80. The latter, normally a stimulating factor, acts paradoxically as an inhibitor of T cells when combined with PDL1 (2).
An interesting fact about PD1 is that it is more broadly expressed than CTLA4. It can be induced on other cell types including B cells and Natural Killer cells to limit their activity. Thus, PD1 blockade not only stimulates T cells but also Natural Killer cells lytic activity and antibody production.
Apart from being as revolutionary and promising as anti-CTLA4, the main advantage of PD1 blockade seems to be its reduced degree of immune toxicity, which happens to be one of the major concerns regarding checkpoint inhibition therapy.
Combining strategies
As stated above, the very first clinical studies observed that immune checkpoint inhibition acted better in a setting of a pre-existing anti-tumoral immune response. Apart from the now classic cellular vaccine combination, other “adjuvant” therapies have been proposed, among those are antibodies targeting tyrosine kinase receptors (that are overexpressed in tumors upregulating immune checkpoints), certain chemotherapeutic agents, vascular endothelial growth factor, etc.
However, the most important combination strategy is the combination of both anti-CTLA4 and anti-PD-1.
Hypothesis of the efficiency of such combination was authorized by the fact that the two checkpoints acted at different moments in different sites. Thus, a double inhibition should have a synergistic effect. Indeed, the first results of the trials on metastatic melanoma were in favor of a net superiority of combination therapy upon single- agent therapy (8), it was even superior to chemotherapy + checkpoint, radiotherapy + checkpoint and other immunotherapy + checkpoint combinations (1). However, long-term benefits are yet to be demonstrated. Moreover, toxicity issues are particularly of concern with combination therapy, although these are labeled as “manageable in experienced centers” (8).
Finally, whether the results seen in melanoma patients can be transposed to other cancer types has not yet been fully demonstrated, and toxicity might be different with other tumor types as well.
Resistance
As with every other existing therapy, immune checkpoint blockade is not exempt from a risk of resistance.
The first observations rolled in favor of a binary event regarding the initial response to therapy: either the tumor is responding to treatment or it is primarily resistant. When the tumor was responding, it was usually a long-lasting effect.
Yet, as long-term follow up studies were conducted, relapses started emerging, suggesting the existence of acquired (secondary) resistance.
There is a wide range of causes and mechanisms of both primary and secondary resistance to checkpoint-inhibition therapy. These can be classified into three main categories (3): insufficient generation of anti-tumor T cells, inadequate function of anti-tumor T cells, and impaired formation of T-cell memory.
Insufficient anti-tumor T-cell generation: a weak T-cell generation response can be due to either tumor-intrinsic mechanisms or tumor-extrinsic mechanisms. Intrinsic mechanisms include mainly genetic and epigenetic alterations that influence antigen formation, presentation or processing.
Mutational burden is a particularly important factor; an increased number of mutations leads to enhanced antigen formation and thus stimulating more clones of T-cells. This is the main reason explaining the efficiency of checkpoint-inhibition against tumors harboring high levels of mutational burden (like melanoma) and its inefficiency against tumors that do not. Consistent with that, DNA-mismatch repair deficiencies are associated with better responses, while the absence of mismatch repair deficiencies is associated with resistance.
Apart from that, the disruption of various cellular signaling pathways in T cells can also lead to resistance. Among these, loss of PTEN is one of the most important, particularly in resistance to PD1.
Speaking of PD1 resistance, an Innate PD1 Resistance (IPRES) gene signature has been identified and involves immunosuppressive cytokines, transcription factors and proangiogenic factors.
Finally, extrinsic mechanisms involve the action of stromal or immune non-cancerous cells; as well as complex host-microbial interactions.
Inadequate anti-tumor T-cell function: a dysfunction of T-cells can be due to alteration of key cellular pathways like mutations in JAK1/JAK2 proteins or deletion of IFNγ receptors that are associated with resistance to PD1.
Other mechanisms include upregulation of the receptors of immune-checkpoints CTLA4 and PD1 or use of alternate immune-checkpoints or co-inhibitory receptors, as well as increased formation of suppressive cytokines and recruitment of immune suppressive cells (Tregs or myeloid-derived suppressor cells).
Impaired formation of T-cell memory: the secret of the long-lasting response to checkpoint inhibition is the formation of T-cell memory. Loss of this memory may be involved in secondary resistance. Mechanisms of memory formation are not well understood but may involve specific transcriptional programs, the impairment of which by tumor cells
alters T-cells’ memory.
Hallmarks of response
As more discoveries were made regarding the mechanisms of resistance, models have been proposed to predict a priori how a tumor might respond to the treatment. Cogdill & al propose 4 pillars that insure a response to checkpoint blockade (10).
We will just cite them without digging into the details, may the reader forgive us for that (or thank us, it depends…):
• Tumor genome and epigenome: tumor burden (amounts and types of alterations, etc.), ability of tumor to evade immunity (MHC expression, IFN signaling status, etc.), metabolic factors (glucose, oxidative stress, etc.).
• Tumor microenvironment: ability to exclude immune infiltrate (Wnt/β-catenin), cytokines/soluble factors (VEGF, IL-6, IL-17, TGF-β).
• Immunity status: innate and adaptive immunity capacities, density and phenotype of immune cells, cytokine markers of chronic inflammation, maturation/inhibition of antigen presenting cells.
• Environment’s Internal/external factors: soluble factors affecting host responses (stress hormones, glucocorticoids), antigenic simulation and tolerance.
Adverse effects
Checkpoint inhibitors play a crucial role in regulating immune responses and maintaining self-tolerance. It is not surprising then that checkpoint blockade induces various immune-related adverse effects that closely resemble autoimmune disease (but hopefully lack chronicity). Some studies report a frequency of over 60% in patients; in the vast majority, these are mild (11).
General signs: fatigue is one of the most common side effect, but remains mild in the majority of patients. Fever and chills have also been described but are rare. These appear to be related to the increased inflammation resulting from treatment.
Allergic reactions have been reported somewhat frequently but were rarely severe or life-threatening.
Dermatologic toxicity: it is the most common immune-related adverse event, occurring in approximately 50% of patients. It is also the earliest, with an average onset of 3 to 4 weeks after treatment initiation.
Manifestations are usually mild like rash or pruritus but can be rarely as dangerous as a Stevens-Johnson syndrome.
Pathology studies report lymphocytic infiltrates extending into the skin which points to an autoimmune mechanism.
Particularly, vitiligo, which can be observed in up to 10% of patients with melanoma, is due to the development of anti-melanA T cells. Interestingly, the development of vitiligo can be a good predictor for a durable response
against melanoma (12).
Gastrointestinal toxicity: second most common adverse effect , in which the chief complaint is diarrhea. Its presence is suggestive of the presence of inflammation, usually a colitis, but the fact that diarrhea can be present without underlying inflammation suggests that other mechanisms may be involved (13). Diarrhea is more frequent with anti-CTLA4 and appears to be dose-dependent.
Colitis related to immune-checkpoint inhibition is probably due to loss of tolerance. But here again other mechanisms may be involved, including intestinal dysbiosis or pharmacokinetic/pharmacodynamic characteristics
of the drugs, which could explain the wide range of variations in clinical manifestations.
Interestingly, these colitises share several clinical, histological and endoscopic similarities with inflammatory bowel disease, which raises the question of these drugs’ safety in patients with history of inflammatory bowel disease. Up to now, the question was not properly studied.
Hepatotoxicity: it is not a rare event in patients treated with immune-checkpoint inhibitors but remains asymptomatic in most cases, revealing itself only by a biological elevation in liver enzymes.
As we already know that T cells play a major role in the pathogenesis of classic autoimmune hepatitis, it was speculated that the mechanism of the latter is the same as the one associated with checkpoint-inhibition. However, serologic markers of autoimmune hepatitis are negative in cases of checkpoint-inhibition toxicity, challenging thus the theory. Histopathological and molecular features still need to be deeply studied in order to figure out the
mechanism of this toxicity (14).
Pneumonitis: it is an uncommon but potentially severe or fatal complication of this treatment. It is not well studied but appears to manifest by common symptoms like dyspnea or coughing, even though asymptomatic patients are common.
There are no characteristic radiographic or pathologic features for checkpoint inhibition lung toxicity. In fact it remains a diagnosis of exclusion. The presence of any symptoms of pneumonitis warrants interruption of immune- checkpoint delivery and initiation of steroid treatment (12).
Endocrinopathies: the thyroid and pituitary glands are most commonly targeted by auto-antibodies in the setting of checkpoint blockade.
Autoimmune thyroid disease can manifest either as hypothyroidism or hyperthyroidism (less common).
Hypophysitis’s manifestations are non-specific like fatigue or headache and the diagnosis is done on the basis of laboratory findings. Hypophysitis is more frequent with anti-CTLA4, which are IgG1 antibodies, than with anti PD1, which are IgG4 antibodies. Knowing that IgG1 can activate the classical complement pathway, it was hypothesized that the latter might be a possible mechanism of anti-CTLA4 related hypophysitis (15). Anti-PD1 related hypophysitis on the other hand appears to be mainly conditioned by genetic predispositions.
Adrenal insufficiency related to checkpoint inhibition, a very rare but most critical event, is an emergency that can cause severe dehydration, hypotension and electrolyte imbalances.
Type 1 diabetes mellitus cases have also been reported (11).
Conclusion
Although truly revolutionary, immune-checkpoint inhibition appears to be a complex issue and the multiple challenges that it raises should be properly discussed in our country in order to ensure an adequate benefit without harming patients.
Balancing their benefits and disadvantages may seem difficult but remains less challenging compared to the overall toxicity observed in chemotherapy. Nonetheless, it seems to be lightening the load of an already burdensome
disease and could be the starting point of a definitive solution. Either way, a more targeted version of this treatment remains to be discovered.
A discovery worthy of a future Nobel prize?
References
1- Park et al. Future prospects of immune checkpoint blockade in cancer: from response prediction to overcoming resistance. Experimental & Molecular Medicine (2018)50:109.
2- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. 10.1038/nrc3239
3- Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118:9–16.
4- Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al: A new member of the immunoglobulin superfamily – CTLA-4. Nature 1987; 328:267–270.
5- Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA 4 blockade. Science 271, 1734–1736 (1996).
6- van Elsas, A., Hurwitz, A. A. & Allison, J. P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA 4) and granulocyte/macrophage colonystimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 190, 355–366 (1999).
7- Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
8- Haanen J. B., Robert C. Immune checkpoint inhibitors. Progress in Tumor Research. 2015;42:55–66.
9- Ishida Y, Agata Y, Shibahara K, Honjo T: Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11:3887–3895.
10- Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer. 2017;117:1–7. doi:10.1038/bjc.2017.136.
11- Michael Postow, Jedd Wolchok. Patient selection criteria and toxicities associated with checkpoint inhibitor immunotherapy (2018). Sadhna R Vora (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
12- Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev. 2016;44:51–60.
13- Samaan MA, Pavlidis P, Papa S, Powell N, Irving PM. Gastrointestinal toxicity of immune checkpoint inhibitors: from mechanisms to management. Nat Rev Gastroenterol Hepatol (2018) 15:222–34.10.1038/nrgastro.2018.14.
14- Reddy HG, Schneider BJ, Tai AW. Immune Checkpoint Inhibitor-Associated Colits and Hepatitis. Clin Transl Gastroenterol. 2018 Sep; 9(9): 180.
15- Byun D.J., Wolchok J.D., Rosenberg L.M., Girotra M. Cancer immunotherapy – immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. 2017;13:195–207.