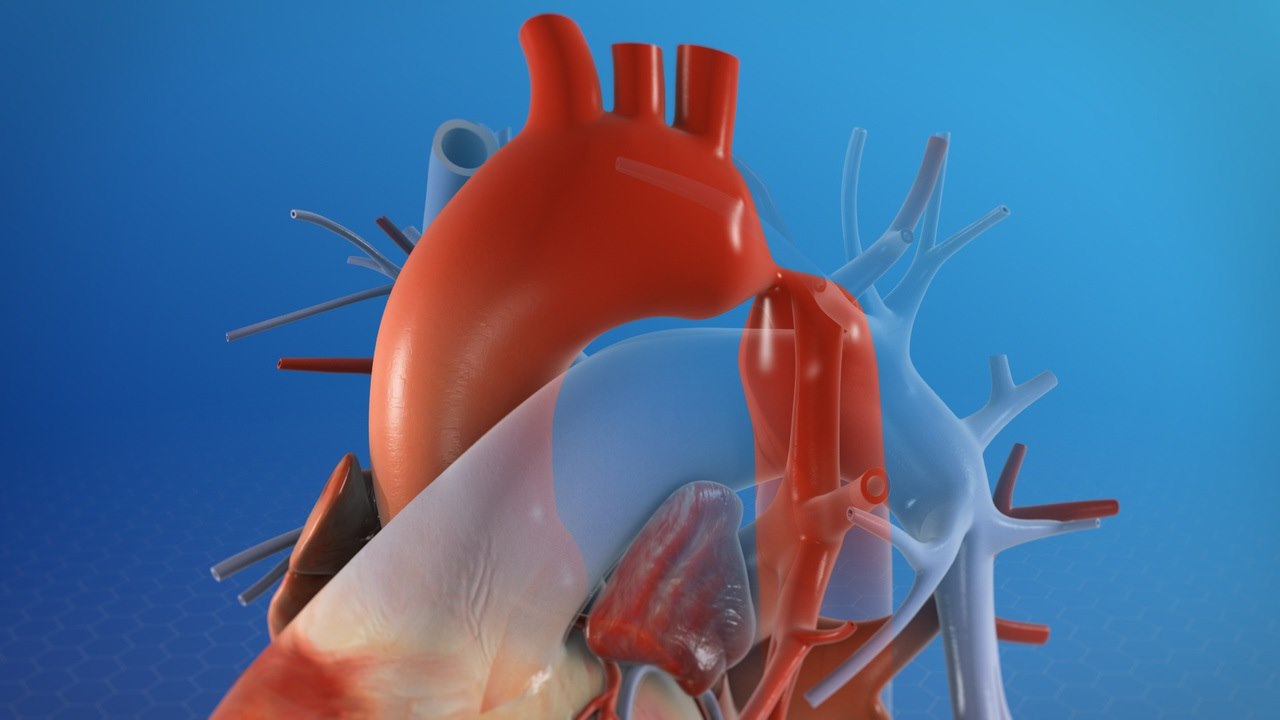
Coarctation Aortique
Khadidja AYADI
La coarctation aortique est une maladie cardiaque congénitale fréquente, elle est souvent associée à d’autres malformations cardiaques et vasculaires. Les patients souffrant d’une coarctation aortique se présentent généralement avec des pouls fémoraux faibles, voire imperceptibles, une différence de pression artérielle plus de 20 mmHg entre les membres supérieurs et inférieurs, ou un souffle typique à l’auscultation cardiaque. Le diagnostic de la coarctation est basé sur des méthodes d’imagerie non invasive, essentiellement l’échographie. Le traitement habituel de la coarctation aortique est chirurgical. Cependant, même après une opération réussie, des données indiquent la présence d’une morbidité et d’une mortalité cardiovasculaires accrues à l’âge adulte, nécessitant un suivi permanant et au long cours des patients traités.
Introduction
Coarctation vient du terme latin « coarctio » qui signifie littéralement un rapprochement. Dans le contexte d’une malformation cardiaque congénitale, la coarctation décrit le plus souvent une zone de rétrécissement de l’aorte thoracique dans la région de l’insertion du canal artériel avec ou sans malformations supplémentaires de la crosse aortique.1 La coarctation aortique est la cinquième malformation cardiaque congénitale la plus commune.3 Elle représente 4 à 6% de toutes les malformations cardiaques congénitales avec une prévalence de 4 cas pour 10.000 naissances.14 Elle est plus fréquente chez les hommes que chez les femmes.1
Historique
La première description de la coarctation aortique est généralement attribuée à Johann Freidrich Meckel qui a présenté un cas chez une femme de 18 ans à l’Académie Royale des Sciences de Berlin en 1750. Certains auteurs évoquent toutefois le rôle de Morgagni dans la découverte de cette pathologie. En 1791, un deuxième cas de coarctation aortique (CoA) a été présenté par monsieur Paris, qui décrivit l’autopsie d’une femme âgée de 50 ans dont les artères thoraciques étaient plus épaisses et plus tortueuses que la normale, et dont la partie de l’aorte qui se trouve en aval de la crosse aortique, entre le ligament artériel et le premier espace intercostal inférieur, était si rétrécie qu’elle avait une épaisseur d’une plume d’oie. En revanche, la partie distale du vaisseau était de calibre normal. La dissection n’a révélé aucune cause ni dans l’aorte ni dans sa vascularisation à laquelle cette condition inhabituelle pouvait être attribuée.1
Etiologie
La pathogenèse exacte de la CoA congénitale est inconnue, cependant trois théories ont été suggérées pour expliquer les anomalies de l’arc aortique.1,14 Selon une première théorie, la CoA serait due à l’extension du tissu canalaire dans la paroi de l’aorte thoracique du foetus, souvent sous la forme d’anneaux qui entourent presque complètement le vaisseau. Cette théorie est étayée par plusieurs études où il a été observé un soulagement de la CoA après l’administration de la prostaglandine E1, qui permet de maintenir ouvert le canal artériel, suggérant ainsi, une extension fonctionnelle du tissu canalaire.3,17 Une deuxième hypothèse évoque le rôle d’une réduction du flux sanguin antérograde passant par l’aorte ascendante qui conduit à un développement anormal de la crosse aortique chez le foetus. Cette réduction serait possiblement due à une légère réorientation de l’angle de jonction du canal artériel avec l’aorte, dirigeant ainsi le flux sanguin du canal artériel directement vers l’aorte descendante, avec pour résultat une diminution du flux sanguin dans l’aorte ascendante.17 Enfin, une dernière théorie propose une implication d’anomalies du développement de la crête neurale dans la genèse de la CoA. En effet, la crête neurale donne naissance à des structures cardiaques telles que les artères carotides, l’arc aortique et le canal artériel qui se forment respectivement à partir du IIIe, IVe et VIe arcs pharyngiens. Selon Kappetein et al., des facteurs génétiques et environnementaux entraineraient des atteintes des cellules de la crête neurale avant ou au cours du développement des arcs IV et VI, et pourraient expliquer l’éventuelle présence d’anomalies dans les structures qui se développent à partir de ces arcs, notamment le canal artériel et l’arc aortique, et donc, la formation de la CoA.13 Il convient de noter que d’autres facteurs génétiques ont été impliqués dans la pathogénèse de la CoA. Freylikhman et al., ainsi que d’autres auteurs, ont observé une mutation du gène NOTCH1 chez les patients souffrant de coarctation aortique, mettant ainsi en exergue le rôle de la signalisation NOTCH1 dans le développement du système cardiaque.11 En outre, une prévalence de CoA de 20% a été observée dans le syndrome de Turner, avec une incidence plus élevée chez les sujets atteints de monosomie X (45XO).7 Il est à noter que, la CoA peut être d’origine acquise, due à des maladies inflammatoires de l’aorte, telles que l’artérite de Takayasu, ou, rarement, une athérosclérose sévère.14
Morphologie
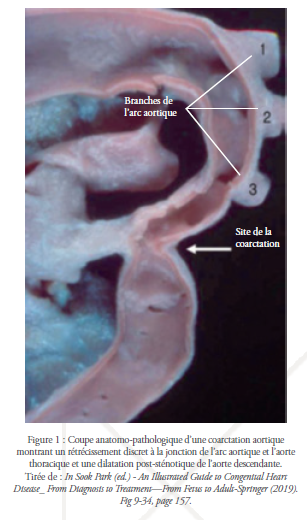
La CoA n’est pas une entité uniforme, elle représente un spectre de rétrécissements de la crosse aortique allant d’une forme discrète, localisée, affectant un site précis de la crosse aortique, à l’hypoplasie tubulaire, qui affecte un long segment de la crosse aortique. Les deux formes peuvent coexister. L’interruption aortique représente la forme la plus grave du spectre.19,10 La CoA peut être isolée, mais elle est souvent associée à d’autres lésions cardiovasculaires qui se produisent majoritairement à proximité de la jonction de l’arc aortique et du canal artériel, notamment l’hypoplasie de la crosse aortique, la sténose sous-aortique, les anomalies de la valve mitrale, les communications interventriculaire ou interatriale, la persistance du canal artériel ou, plus fréquemment, une bicuspidie valvulaire aortique (50-75%),5 qui peut se compliquer tardivement par des sténoses calciques, une insuffisance aortique ou un risque accru d’endocardite infectieuse.1
Physiopathologie
Durant la vie foetale et jusqu’à la naissance la coarctation aortique ne pose pas de problèmes hémodynamiques car le sang éjecté par le coeur passe par le canal artériel dans l’aorte thoracique descendante, en contournant le site de rétrécissement pour rejoindre la circulation systémique. Pendant la période néonatale ; lorsque le canal artériel et le foramen ovale commencent à se fermer, le débit cardiaque qui doit traverser le segment aortique rétréci augmente progressivement, provoquant des modifications hémodynamiques pouvant aller d’une légère hypertension systolique à une insuffisance cardiaque selon la gravité de la coarctation et l’éventuelle présence d’autres lésions associées.14 Aussi, le flux diastolique dans les artères coronaires diminue à mesure que le tension de la paroi du ventricule gauche augmente, conduisant à une ischémie, en particulier du sous-endocarde.1
A l’occasion de la diminution du débit cardiaque, une hypoperfusion périphérique provoque l’utilisation tissulaire des voies métaboliques anaérobies, augmentant la production de lactates et conduisant finalement à une acidose métabolique.1
Si l’obstruction est légère, des mécanismes de compensation peuvent se mettre en place, principalement sous la forme d’une hypertrophie du ventricule gauche.1
Aussi, durant l’enfance une circulation collatérale se développe progressivement, contournant l’obstruction pour augmenter la perfusion vers les extrémités du corps. Souvent, une volumineuse artère aberrante nait de l’artère sous-clavière droite pour alimenter l’aorte sous la coarctation, ainsi que diverses branches de l’artère sous-clavière gauche, y compris le tronc thyrocervical et les artères intercostales gauches via l’artère thoracique interne gauche.1
Les enfants plus âgés et les adultes développent au niveau du site de coarctation un épaississement fibreux de l’intima avec des couches concentriques de collagène et des degrés variables d’élastine et des cellules musculaires lisses, ainsi qu’une médianécrose kystique et une dilatation post-sténotique.1 Un thrombus peut se former au niveau de la coarctation, ce dernier aggrave la sténose causée par la prolifération intimale et peut conduire à l’oblitération presque complète de la lumière aortique ; ainsi, toute la perfusion distale devient dépendante de la circulation collatérale.1 Des complications à distance ont également été décrites, notamment l’anévrisme sacculaire du cercle de Willis et d’autres pathologies secondaires à l’hypertension artérielle.1
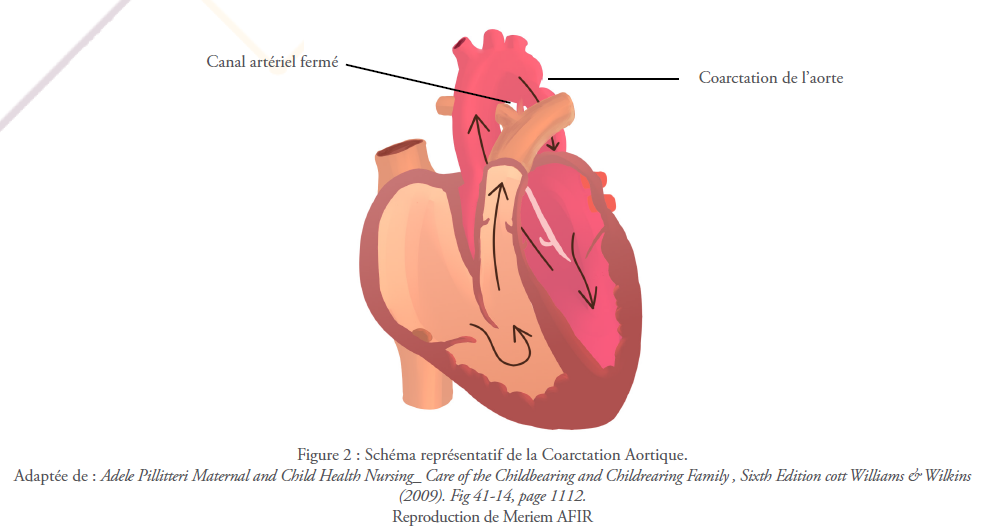
Symptomatologie
Chez les nourrissons :
La symptomatologie commence à se manifester environ une semaine après la naissance, notamment par une mauvaise alimentation et un retard de croissance, des sueurs profuses ainsi qu’une dyspnée. À l’examen physique, un signe particulièrement évocateur est l’absence ou le retard du pouls fémoral, avec des pouls conservés aux membres supérieurs.1
En outre, des anomalies à l’auscultation cardiaque peuvent être retrouvées, témoignant de pathologies associées tels que des signes en rapport avec une persistance du canal artériel, une sténose aortique ou une communication interventriculaire.14 Les nouveau-nés présentant une CoA associée à une persistance du canal artériel avec un shunt droit-gauche au niveau de l’aorte thoracique descendante peuvent présenter une cyanose différentielle (cyanose au niveau des membres inférieurs seulement alors que les membres supérieurs sont normalement colorés).14
Les nouveau-nés atteints d’une CoA sévère peuvent également présenter des degrés variables d’insuffisance cardiaque et/ou un état de choc à la fermeture du canal artériel.1
Chez les enfants plus âgés et les adultes :
La majorité des patients atteints d’une légère CoA sont asymptomatiques grâce au développent précoce et important de la circulation collatérale.1
À l’examen clinique, une hypertension systémique est constatée, avec plus de 20 mmHg de différence de pression entre les membres supérieurs et inférieurs. Le défaut d’irrigation des membres inférieurs se manifeste également par des pouls fémoraux faibles voire imperceptibles, ou d’une claudication douloureuse à l’effort.
À l’auscultation cardiaque, on retrouve également un souffle de coarctation aortique typique. Il est le plus audible au niveau de la fosse infra-claviculaire gauche et irradie vers l’arrière vers la scapula gauche. Il est continu, avec un pic télé-systolique et se continue en début de la diastole, ce qui correspond à la queue diastolique, également observée à l’échographie Doppler. Des souffles continus supplémentaires peuvent être générés par les artères collatérales.1 On peut également retrouver des épistaxis ou des céphalées qui sont dues à l’hypertension systémique.1,14
Plus rarement, les patients se présentent avec des complications sévères de la maladie hypertensive telles que des hémorragies sous-arachnoïdiennes ou une rétinopathie hypertensive.1 Plus tard dans la vie, un risque accru de maladie coronarienne a été rapporté.1 L’insuffisance cardiaque est rare entre 1 et 30 ans, bien qu’elle se développe chez environ 65% des patients qui survivent plus de 40 ans.2
Diagnostic Prénatal
Le diagnostic de la CoA peut être établi dès le premier trimestre de la gestation, bien que les méthodes usuelles de dépistage ne soient pas très fiables, si bien que de nombreux cas ne sont détectés qu’après la naissance. 10 Le dépistage prénatal de la CoA est associé à un nombre élevé de faux positifs, surtout en fin de gestation.5 Plusieurs marqueurs échocardiographiques peuvent améliorer les taux de détection prénatale, notamment le diamètre de l’isthme aortique, le diamètre du canal artériel, le rapport isthme/ducal, les scores Z dérivés des mesures de l’isthme aortique distal et du canal artériel, la présence du « shelf » (indentation de la paroi postérieure de l’isthme aortique) et la perturbation du flux isthmique.15
Diagnostic Postnatal
Le diagnostic clinique de la CoA est basé sur la présence d’une hypertension artérielle, avec plus de 20 mmHg de différence de pression entre les membres supérieurs et inférieurs et la diminution du pouls fémoral. Le diagnostic est confirmé par des méthodes d’imagerie non invasive essentiellement l’échocardiographie.14
L’échocardiographie : C’est la méthode de diagnostic la plus répandue chez les enfants atteints d’une anomalie de la crosse aortique. L’échocardiographie montre un rétrécissement segmentaire juste en-dessous de l’artère sous-clavière gauche ; il peut aussi y avoir un segment plus long de rétrécissement au niveau de l’isthme.14 En absence de persistance du canal artériel, la sévérité hémodynamique de la coarctation peut être facilement évaluée par l’écho-Doppler.1, 14 L’échocardiographie peut également détecter les anomalies cardiaques associées, y compris l’hypoplasie aortique, et peut être utilisée pour le suivi après réparation.14
L’IRM cardiaque : Bien que l’IRM soit limitée chez les petits enfants en raison de la nécessité d’une sédation ou d’une anesthésie, elle constitue un excellent outil pour évaluer la réparation postopératoire et la crosse aortique chez les adultes diagnostiqués tardivement.1 C’est la méthode privilégiée pour l’évaluation du traitement et des complications de la coarctation aortique. Elle peut également quantifier le flux collatéral, révéler des complications de la CoA et fournir des détails sur la masse et la fonction du ventricule gauche.1
L’angioscanner : Bien que l’IRM soit généralement préférée pour diminuer l’exposition aux rayonnements lors d’examens répétés,1 l’angioscanner est plus largement disponible. Il assure également des temps de numérisation plus courts et une meilleure résolution d’image. En outre, l’angioscanner est la méthode recommandée chez les patients qui ont des endoprothèses vasculaires.14
La radiographie thoracique : Chez les nourrissons une radiographie thoracique standard révèle une cardiomégalie. Chez les enfants les plus âgés la cardiomégalie peut se présenter suite à une hypertrophie du ventricule gauche due à l’hypertension artérielle. En outre, les signes les plus caractéristiques chez ces patients sont le signe du 3 sur le bord gauche du médiastin, suite à une dilatation post-sténotique de l’aorte, et l’encoche costale, généralement observée à partir de 4 à 6 ans. A l’âge adulte, environ 75% des patients présentent une encoche des côtes souvent dans le tiers médian des bords inférieurs de la 4e à la 8e côte, suite à la dilation des artères intercostales collatérales.1
Le cathétérisme cardiaque : Il est généralement effectué en association avec une intervention thérapeutique chez les enfants présentant une CoA isolée. Chez l’adulte, le cathétérisme cardiaque est indiqué lorsqu’on suspecte une coronaropathie associée.14
Prise en Charge
Stabilisation :
L’objectif initial est de maintenir la stabilité hémodynamique et ce en maintenant le canal artériel ouvert via l’administration de prostaglandine E1 (PGE1).1 La PGE1 doit être administrée jusqu’à ce qu’un diagnostic ou un traitement définitif soit établi.16
Bien qu’il n’y ait pas d’essais contrôlés randomisés, une amélioration occasionnelle à l’administration de la PGE1 a été documentée chez des nourrissons de 5 semaines.1 L’efficacité est généralement moins impressionnante chez les nouveau-nés plus âgés et chez les personnes dont le canal est fermé à la présentation.1 En revanche, l’administration de la PGE1 peut être associé à une dyspnée nécessitant une ventilation mécanique, une hypotension, une sensibilité accrue aux infections, une diarrhée et plus rarement une coagulopathie.1
Correction de la CoA :
Tout patient présentant une coarctation sévère devrait, une fois celle-ci stabilisée, bénéficier d’une chirurgie correctrice. Les indications de la chirurgie après la naissance sont une hypertension systémique, une circulation collatérale importante et/ou une coarctation sévère impliquant une réduction de plus de 50% du diamètre de la lumière.1,16
La décision de procéder à une réparation chirurgicale ou à une intervention par cathéter est basée sur plusieurs facteurs, notamment l’âge, l’intervention précédente et la complexité de l’anatomie.1 Il existe plusieurs techniques chirurgicales et interventionnelles possibles.
Parmi les techniques chirurgicales nous citons :
Résection anastomose : elle est utilisée généralement pour réparer la coarctation aortique discrète dont les résultats sont associés à un faible taux de mortalité et une faible incidence de recoarctation ou de formation d’anévrisme.1
Aortoplastie-patch sous-clavière : elle est indiquée chez les nourrissons atteints de CoA à long segment.16
Tube aorte aortique latéral : c’est une option chirurgicale intéressante pour les patients adultes présentant une obstruction récurrente de l’arc aortique, afin d’éviter une dissection trop importante et des complications postopératoires.18
Aortoplastie par patch : elle est généralement évitée en raison de l’apparition fréquente d’anévrisme aortique.16
La recoarctation est la complication majeure qui peut survenir à long terme après la réparation chirurgicale de la coarctation, en particulier chez les nouveau-nés et les jeunes enfants.16 Jusqu’à présent, aucune opération ne semble avoir une nette supériorité.2
Aussi, parmi les techniques interventionnelles figurent :
Angioplastie par ballonnet : une petite étude randomisée a montré que l’angioplastie par ballonnet utilisée pour traiter les enfants atteints de coarctation était associée à une incidence plus élevée de formation d’anévrisme et de lésions artérielles ilio-fémorales par rapport à la chirurgie.1 Cependant, elle peut être utile chez les nouveau-nés qui sont de très mauvais candidats à la chirurgie en offrant une période d’optimisation médicale avant l’opération.1 En général, l’angioplastie par ballonnet est plus efficace pour la coarctation récurrente que pour la CoA primaire, avec une incidence plus faible de formation d’anévrisme.1
Angioplastie par stent : elle est efficace chez les enfants présentant une coarctation primaire ou récurrente, même chez les patients pesant moins de 20 kg. Néanmoins, chez les jeunes enfants, le risque de détérioration de l’artère fémorale est plus élevé et une nouvelle intervention sera probablement nécessaire pour poursuivre l’expansion du stent. Le succès immédiat de l’implantation d’un stent est extrêmement bon, mais les résultats à long terme de cette intervention sont actuellement moins certains.1
Bien que la chirurgie demeure la thérapeutique de choix pour la prise en charge des patients atteints de CoA, à l’âge adulte, l’angioplastie par ballonnet avec ou sans stent s’impose de plus en plus comme une méthode préférable.1
Complications
Bien que la chirurgie représente la méthode de choix pour la correction de la CoA,12 des complications éventuelles ont été constatées à court terme ainsi qu’à long terme après l’intervention ; en particulier lorsqu’elle est pratiquée tard après la naissance.10
Les complications à court terme comprennent généralement une dissection des tissus autour du site de coarctation, des saignements importants des lignes de suture, un endommagement du canal thoracique ou la dissection extensive des tissus lymphatiques, une hypertension postopératoire, ainsi qu’une paralysie du nerf laryngé récurrent ou du nerf phrénique, le syndrome de Claude Bernard-Horner, et souvent une lésion de la moelle épinière avec une paraplégie consécutive.1
À long terme, après la réparation de la coarctation, l’hypertension est considérée comme l’une des complications les plus courantes et les plus sévères,1 avec une prévalence de 6 à 10% après une réparation néonatale, et 33% et 60% après une réparation à l’âge de 14 et 12 ans, respectivement.6,1
L’origine de l’hypertension artérielle après la correction de la coarctation, est considérée comme multifactorielle.10
Les données suggèrent que malgré une réparation précoce de la CoA pendant l’enfance et en absence de rétrécissement aortique résiduel,10 des modifications des propriétés fonctionnelles et structurelles au niveau du lit artériel précoarctique, avec préservation des artères postcoarctiques seraient un facteur de risque d’hypertension artérielle.10, 3 Certaines études pointent du doigt l’implication du système rénineangiotensine- aldostérone, consécutif à une hypoperfusion rénale et une ischémie rénale,10 comme ayant un rôle majeur dans le développement de l’hypertension à long terme ; cependant, d’autres études ne semblent pas partager cette conclusion.6
D’autres complications peuvent apparaître à long terme après la réparation de la CoA, notamment la recoarctation et l’anévrisme aortique qui sont probablement liés à la technique utilisée lors de l’intervention.1
Esperance de Vie
L’augmentation de l’âge au moment de la réparation et l’hypertension artérielle préopératoire sont deux facteurs de diminution de la durée de vie à long terme.1 Les taux de survie après la correction sont variables. L’étude de Campbell a décrit une cohorte de 181 patients avec un taux de mortalité de 25% chez les patients âgés de plus que 20 ans, 50% avant l’âge de 32 ans, 75% avant 46 ans et 92% avant 60 ans.4 Une autre cohorte de 151 patients atteints de coarctation simple a montré un taux de survie de 98% à 40 ans, 98% à 50 ans et 89% à 60 ans après la réparation de la CoA.9 Cependant, chez les patients atteints de CoA non traitée, l’âge moyen du décès est d’environ 34 ans.8
Conclusion
La coarctation est, souvent, considérée comme un trouble permanent.4 Tous les patients atteints de CoA, réparée ou non, devraient bénéficier d’un suivi et d’une imagerie cardiologique tout au long de leur vie en raison des risques cardiovasculaires à long terme et de la nécessité éventuelle d’une réintervention. Les patients qui ont subi une intervention chirurgicale ou une intervention par trans-cathétérisme pour la CoA devraient être suivis cliniquement au moins une fois par an.16
Références
1- Wernovsky, G., & Anderson, R. (2019). Anderson’s pediatric cardiology (4th ed.).
2- Abbruzzese, P. A., & Aidala, E. (2007). Aortic coarctation: an overview. Journal of cardiovascular medicine (Hagerstown, Md.), 8(2), 123–128.
3- Yokoyama, U., Ichikawa, Y., Minamisawa, S., & Ishikawa, Y. (2017). Pathology and molecular mechanisms of coarctation of the aorta and its association with the ductus arteriosus. The journal of physiological sciences : JPS, 67(2), 259–270.
4- Horlick, E. M., McLaughlin, P. R., & Benson, L. N. (2007). The adult with repaired coarctation of the aorta. Current cardiology reports, 9(4), 323–330.
5- Dijkema, E. J., Leiner, T., & Grotenhuis, H. B. (2017). Diagnosis, imaging and clinical management of aortic coarctation. Heart (British Cardiac Society), 103(15), 1148–1155.
6- Kenny, D., Polson, J. W., Martin, R. P., Paton, J. F., & Wolf, A. R. (2011). Hypertension and coarctation of the aorta: an inevitable consequence of developmental pathophysiology. Hypertension research : official journal of the Japanese Society of Hypertension, 34(5), 543–547.
7- Wong, S. C., Cheung, M., & Zacharin, M. (2014). Aortic dilatation and dissection in Turner syndrome: what we know, what we are unclear about and what we should do in clinical practice?. International journal of adolescent medicine and health, 26(4), 469–488.
8- Golden, A. B., & Hellenbrand, W. E. (2007). Coarctation of the aorta: stenting in children and adults. Catheterization and Cardiac Angiography & Interventions, 69(2), 289–299.
9- Choudhary, P., Canniffe, C., Jackson, D. J., Tanous, D., Walsh, K., & Celermajer, D. S. (2015). Late outcomes in adults with coarctation of the aorta. Heart (British Cardiac Society), 101(15), 1190–1195.
10- Vigneswaran, T. V., Sinha, M. D., Valverde, I., Simpson, J. M., & Charakida, M. (2018). Hypertension in Coarctation of the Aorta: Challenges in Diagnosis in Children. Pediatric cardiology, 39(1), 1–10. Références
11- Freylikhman, O., Tatarinova, T., Smolina, N., Zhuk, S.,Klyushina, A., Kiselev, A., Moiseeva, O., Sjoberg, G., Malashicheva, A., & Kostareva, A. (2014). Variants in the NOTCH1 gene in patients with aortic coarctation. Congenital heart disease, 9(5), 391–396.
12- Hornung, T. S., Benson, L. N., & McLaughlin, P. R. (2002). Interventions for aortic coarctation. Cardiology in review, 10(3), 139–148.
13- Kappetein, A. P., Gittenberger-de Groot, A. C., Zwinderman, A. H., Rohmer, J., Poelmann, R. E., & Huysmans, H. A. (1991). The neural crest as a possible pathogenetic factor in coarctation of the aorta and bicuspid aortic valve. The Journal of thoracic and cardiovascular surgery, 102(6), 830–836.
14- Agarwala BN, Bacha E, Cao Q, Hijazi MZ. Clinical manifestations and diagnosis of coarctation of the aorta. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com
15- Buyens, A., Gyselaers, W., Coumans, A., Al Nasiry, S., Willekes, C., Boshoff, D., Frijns, J. P., & Witters, I. (2012). Difficult prenatal diagnosis: fetal coarctation. Facts, views & vision in ObGyn, 4(4), 230–236.
16- Agarwala BN, Bacha E, Cao Q, Hijazi MZ. Management of coarctation of the aorta. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com
17- Rudolph, A. M., Heymann, M. A., & Spitznas, U. (1972). Hemodynamic considerations in the development of narrowing of the aorta. The American journal of cardiology, 30(5), 514–525.
18- Shinkawa, T., Chipman, C., Holloway, J., Tang, X., Gossett, J. M., & Imamura, M. (2017). Single center experience of aortic bypass graft for aortic arch obstruction in children. Heart and vessels, 32(1), 76–82.
19- Matsui, H., Adachi, I., Uemura, H., Gardiner, H., & Ho, S. Y. (2007). Anatomy of coarctation, hypoplastic and interrupted aortic arch: relevance to interventional/surgical treatment. Expert review of cardiovascular therapy, 5(5), 871–880.
20- Vigneswaran, T. V., Sinha, M. D., Valverde, I., Simpson, J. M., & Charakida, M. (2018). Hypertension in Coarctation of the Aorta: Challenges in Diagnosis in Children. Pediatric cardiology, 39(1), 1–10.