Résistance à l’Insuline
Amine LABOUDI
L’insuline est l’hormone anabolique capitale chez tout être humain. C’est une véritable policière de circulation pour la simple et unique raison qu’elle assure un contrôle minutieusement mesuré de la disposition énergétique dans l’organisme. Toutefois, l’orchestre des mécanismes régulateurs qu’elle mène peut être perturbé par des carambolages occasionnés entre plusieurs entités physiopathologiques, ce qui engendrerait potentiellement l’émergence d’une désobéissance organique aux ordres commandés par cette hormone, vous l’aurez compris, c’est la résistance à l’insuline. Tout au long de cet article, nous allons aborder la globalité des notions fondamentales encadrant la signalisation cellulaire initiée par l’insuline, puis la procédure par laquelle s’installe la résistance à son effet, nous finirons par une simple revue des moyens pharmacologiques que présente le volet thérapeutique du diabète de type II, dont la perte de la sensibilité de l’insuline est de facto le premier mécanisme inculpé.
Introduction
Bien qu’une stratégie de lutte universelle soit mise en place contre le diabète non insulinodépendant, cette maladie ne cesse de prendre de l’ampleur et d’atteindre des proportions épidémiques en terme de mortalité.1,2 Le facteur primo-délinquant dans la pathogenèse de cette affection est la résistance à l’insuline, dont la sécrétion devient disproportionnée.3
L’insuline maintient, si cela se dit, une certaine homéostasie d’énergie en facilitant l’entrée de glucose dans les tissus périphériques et en diminuant la libération des lipides stockés au niveau des tissus adipeux.4 Cette homéostasie, finement préservée, peut subir des déséquilibres dans le cas où la sécrétion ou l’efficacité de l’insuline soient réduites.5
Le thème principal de l’article se pose sur la résistance à l’insuline, qui constitue l’éventualité physiopathologique clé dans le développement du diabète de type II.2 On y trouve une réponse biologique détériorée aux stimuli apportés par l’insuline, ce qui mène l’organisme à augmenter sa production de façon qu’une hyperinsulinémie compensatrice soit mise en place. Par conséquent, on observe l’apparition d’une hyperglycémie, d’une dyslipidémie et d’une hypertension, toutes dans le cadre d’un syndrome métabolique élargi caractéristique de la pathologie. Cela revient au fait que même si l’effet hypoglycémiant de cette hormone soit mis sous une relative suppression, l’insuline garde son rôle de pilote de la lipogenèse, indépendamment des circonstances, et donc toute hyperinsulinémie réactionnelle résultante de la résistance, qui s’installe à pas doux, mène ces sujets à être plus exposés aux dyslipidémies par rapport à ceux qui en sont indemnes.2, 5, 6
La signalisation intracellulaire de l’insuline, un labyrinthe ardu!
Le corps humain comprend un seul gène codant pour l’insuline, c’est l’INS. Il est localisé au niveau du chromosome 11. Sa transcription est contrôlée par un large panel d’éléments modulateurs, dont on cite : l’IDX 1 (PDX1), le MafA, la neuroD1 et d’autres co-régulateurs.5 Ces derniers contribuent à la régulation de l’expression et de la transcription du gène INS au niveau des cellules b du pancréas, d’où l’insuline provient.5
L’insuline, une fois libérée dans le sang, suit son propre tropisme vers ses récepteurs spécifiques (IR : Insulin Receptors) où elle sera fixée. Il s’ensuit l’initiation d’une cascade de signaux aboutissant à l’activation d’une certaine enzyme «proteine kinase B», également appelée Akt. Cette dernière, porte-parole de l’insuline au niveau intracytoplasmique, fait en sorte que tous les mécanismes assurant la néoglucogénèse, la glycogénolyse, et le catabolisme des lipides soient inhibés.5
En effet, le complexe hormone-récepteur néoformé recrute, au niveau intracellulaire, une phosphoinositide-3-phosphate kinase (PI3K) qui se fait phosphoryler, et donc activer par les substrats intrinsèques des récepteurs de l’insuline (IRS) ayant fixé des résidus de phosphotyrosine lors de l’activation du récepteur. Cela génère finalement des phosphatidyl inositol (3, 4, 5)-triphosphate (PIP3).1, 2, 7
Les PIP3 formés s’engagent dans l’activation de l’enzyme 3-phosphoinositide-dependent protein kinase 1 (PDK 1), qui permet la phosphorylation de l’enzyme Akt au niveau de la thréonine 308, mais cela reste insuffisant pour que l’activation de cette dernière soit complète.2 Ce n’est qu’à ce point qu’interviennent les mechanistics targets of rapamycin complexes 2 (mTORCs2) en induisant sa seconde phosphorylation au niveau de la serine 473. Ainsi, l’Atk demeure totalement fonctionnelle.2, 9, 10
À partir de ce stade, plusieurs sentiers sont désormais disponibles à emprunter, tous menant vers un seul et unique objectif commun, qui est le contrôle de l’homéostasie de glucose et des lipides. Cependant, on peut citer en premier lieu les trois substrats primordiaux de l’Atk: (1) la forkhead box protein 01 (FOX01); (2) la peroxisome proliferator-activated receptor gamma coactivator 1a (PGC1a); (3) et la glycogen synthase kinase 1b (GSK3b).2
Une fois que l’Atk est activée par l’insuline, et comme l’on a cité juste au-dessus, elle induit la phosphorylation de la FOX01. Cette dernière, étant responsable de la mise en marche de la voie de néoglucogenèse, se trouve donc inhibée.11
La synthèse du glycogène, étant une forme de stockage tissulaire du glucose, est favorisée par l’inhibition qu’applique l’Atk sur la GSK3, ou bien même par l’activation de l’enzyme Glycogen Synthase 2 (GYS2) via la glucose 6-phophatase (G6P).2
Encore plus, L’Atk active également les mTORC1, qui à leur tour favorisent la voie de lipogenèse impliquant l’activation des Sterol regulatory element binding proteins 1c (SREBP1c). Ils constituent un acteur fondamental en ce qui concerne l’expression cellulaire des gènes requis pour le contrôle du métabolisme des lipides et du cholestérol. Les SREBP1c permettent également la phosphorylation des G6P, qui activent la glycolyse et la synthèse du glycogène, ce qui permet d’abaisser la glycémie.12 Les mécanismes avec lesquels agit l’insuline au niveau intracellulaire par l’intermédiaire principal de l’Atk restent très complexes, et loin d’être totalement élucidés.
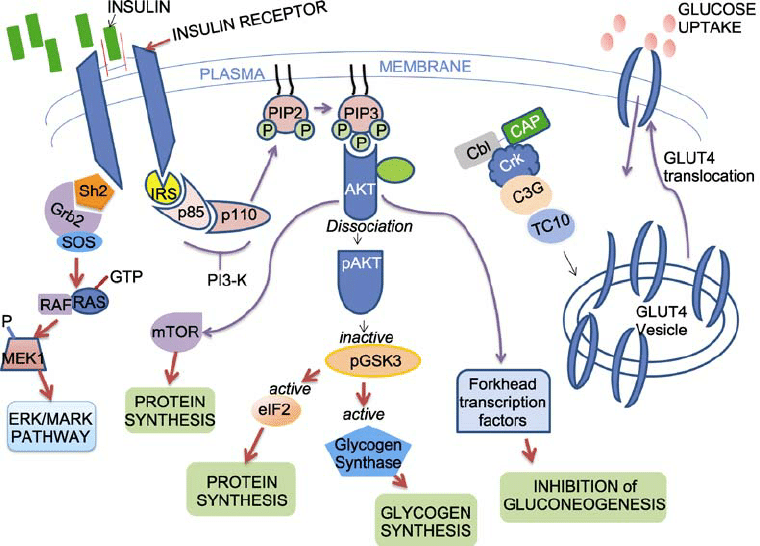
Tirée de : Kumar M, Nath S, Prasad H, Sharma G, Li Y. MicroRNAs: a new ray of hope for diabetes mellitus. Protein & Cell. 2012;3(10):726-738.
La résistance à l’insuline
La résistance à l’insuline se développe à partir de plusieurs interactions complexes allant du génotype de l’individu jusqu’à son mode de vie. En effet, la sensibilité tissulaire à l’insuline dépend de plusieurs agents circulants, incluant les adipokines, les hormones, et les lipides plasmatiques. Ces derniers permettent, à l’état normal, d’ajuster la sensibilité générale à l’insuline vis-à-vis les métabolismes glucidique et lipidique relatifs.13, 14, 15
Les adipocytes réalisent deux fonctions principales, la première, étant la plus basique, consiste en le stockage des gras sous forme de triglycérides, qui sera retransformé en glycérol et acides gras au besoin. La deuxième fonction, moins connue chez le public, est représentée par la sécrétion de certaines hormones, nommées adipokines, dont le rôle majeur est de contrôler l’homéostasie énergétique et de l’insulino-résistance. On en distingue trois principalement, la leptine, l’adiponectine et la résistine. Cela fait du tissu adipeux un organe endocrinien en lui-même, et non pas simplement un organe de stockage comme dixit l’opinion générale.13
Les fonctions des adipokines peuvent être divisées en deux simples catégories, ceux qui stimulent la sensibilité à l’insuline et ceux qui la réduisent. La leptine et l’adiponectine stimulent l’action périphérique de l’insuline.15,16 Il est à préciser, par contre, que le taux de leptine est proportionnel au degré d’adiposité corporelle, alors que c’est l’inverse concernant l’adiponectine.15,16 La leptine favorise également l’état de satiété lorsqu’elle est présente dans les taux physiologiques.13
La famille des adipokines qui, à l’inverse, réduit la sensibilité à l’insuline inclut les TNFa, la résistine, l’IL-6 et bien d’autres. Le taux sécrétoire de ces facteurs inhibiteurs est de croissance proportionnelle avec l’adiposité corporelle.14, 15
Qu’elles soient inhibitrices ou stimulatrices, les adipokines sont, dans les conditions physiologiques, en balance continue. Cependant, certains paramètres peuvent perturber cet équilibre. L’obésité y figure sur la liste comme chef d’orchestre.15
Chez une personne obèse, le taux de leptine augmente alors que celui de l’adiponectine chute. L’hyperleptinémie entraîne un effet paradoxal qui consiste en l’installation d’une auto-insensibilité au niveau des tissus cibles, la somme des effets résultants des variations de ces deux adipokines est représentée par la réduction de la sensibilité organique spécifique à l’insuline.15
Cela ne s’arrête pas ici, les adipocytes sécrètent des facteurs chimioattractifs qui entraînent le recrutement de plus de macrophages au niveau du même tissu, ces derniers, étant des cellules immunitaires, sécrètent des cytokines à l’exemple du TNFa et des interleukines, des cytokines qu’on a classé juste en dessus dans la case de facteurs diminuant la sensibilité de l’insuline. C’est ainsi que la boucle est bouclée, le sujet concerné présentera un appétit plus important surajouté à une hyperinsulinémie réactionnelle vu la résistance qui s’installe.15,17
Il est à préciser que la résistance à l’insuline n’épargne pas les sujets présentant une lipodystrophie, cela pourrait paraître paradoxal, mais en connaissant les mécanismes occultes, on comprend rapidement pourquoi. En effet, la dystrophie touchant le tissu adipeux entraîne une déficience de sécrétion des adipokines, notamment la leptine, ce qui réduit significativement la sensibilité de l’insuline.13, 15
D’autres étiologies peuvent être rapportées à l’installation de cette résistance, on parle particulièrement de mutations touchant les gènes codant pour l’une des protéines intégrées dans le processus de signalisation propre de l’insuline, celles qui ont été détaillées précédemment. Toute dysfonction de l’une des composantes de la voie de signalisation aura son propre degré de répercussions, le plus souvent ce sont les anomalies touchant l’Atk qui sont incriminés, mais cela ne doit pas porter à sous-estimer la fréquence des anomalies concernant les autres gènes impliqués dans un tel processus compliqué, on inclut les gènes codant pour l’IR, le régulateur de glucokinase, et l’IGF-I (Insulin Growth Factor-I), voire même l’IRS, ce qui entraînerait un défaut d’activation de la voie sus-citée des PI3K et ainsi, une résistance au stimulus de l’insuline forgerait son apparition.2,9,13 On incrimine également les régimes riches en sodium, et les étiologies iatrogènes (gluco-corticoïdes, anti-adrénergiques, inhibiteurs de protéase…).2, 13, 18
Même si la base de données par rapport aux étiologies engagées dans le développement de la résistance à l’insuline reste plus ou moins élargie, on n’est toujours pas arrivé à la phase de satiété souhaitée. La génétique qui y est impliquée, en particulier, reste a priori le thème primordial d’actualité scientifique sur ce sujet.
Diabète de type II, un passage sur la thérapeutique
Comme déjà expliqué, la résistance à l’insuline constitue le premier mécanisme physiopathologique définissant le diabète de type II, c’est pour cela que les attitudes thérapeutiques par rapport à ce dernier reposent essentiellement sur des molécules ayant la capacité de jouer sur le fil de la sensibilité tissulaire à l’insuline.
Le schéma thérapeutique est fondé de base sur certaines règles hygiéno-diététiques à suivre, dont on cite : la pratique d’une activité sportive régulière côte à côte avec le respect d’un régime hypocalorique, mais le plus important reste la perte de poids.20 Le but de cette procédure est d’améliorer les fonctions mitochondriales, de diminuer la génération des radicaux libres, d’augmenter l’efficacité de l’insuline selon les mécanisme physiologiques sus-cités et de contrôler la pression artérielle afin d’éviter des complications relatives telles que les micro-angiopathies et les ischémies.19, 20
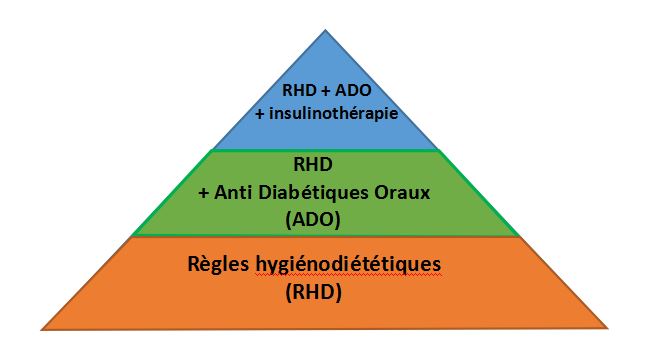
La portion pharmacologique du traitement se base sur des médicaments hypoglycémiants, nécessiteux d’une sécrétion résiduelle de l’insuline, le but étant de la renforcer, et ainsi lui permettre d’atteindre son potentiel maximal. Généralement, une monothérapie est suffisante initialement, mais un second médicament, dont le mécanisme d’action diffère du premier, peut être nécessaire plus tardivement lors d’une seconde phase de thérapie combinée. Vu les essais cliniques limités sur l’éventualité d’usage d’une trithérapie, les recommandations courantes posent la restriction de ne pas dépasser l’association de deux traitements a maxima.19 Il est, cependant, à noter que dans le cas de diabète avancé, l’insuline est indispensable comme composante des deux médicaments constituant la bithérapie prescrite au patient.19
Le choix de la molécule thérapeutique à utiliser chez un sujet donné varie en fonction de plusieurs paramètres, on cite l’âge, le poids, les comorbidités, le stade évolutif du diabète, les préférences du patient… Que cela concerne l’insuline injectable en elle-même ou les antidiabétiques oraux, tels que les sulfonylurées, les glinides, les incrétinomimétiques, et les inhibiteurs de DDP4 nommés aussi les gliptines, tous ces paniers d’options médicamenteuses sont utilisés dans le but de diminuer la glycémie, tout en augmentant la concentration sanguine en insuline.19
Il est cependant à considérer le risque de provoquer une hypoglycémie en administrant d’une manière immesurée les médicaments entraînant des pics de sécrétion d’insuline indépendamment de la concentration de glucose, même s’il arrive que cette dernière soit déjà basse. C’est le cas pour les sulfonylurées, les glinides et bien évidemment l’administration directe d’insuline. Cela peut poser encore plus de complications au long terme étant donné que les taux hautement constants de l’insuline favorisent une lipogenèse continue, ce qui fait gagner du poids additionnel au patient, et alors aggrave sa maladie.19
Certains autres médicaments ne partagent pas ce même effet, leur administration reste dénuée du risque de provoquer de telles complications étant donné la divergence dans les mécanismes d’action entre les différentes classes thérapeutiques. Parmi ces médicaments, on trouve la metformine, qui est recommandée en première intention par la majorité des guidelines de nos jours, à condition que le patient ne présente pas des facteurs de risque vis-à-vis sa prescription, dont on cite principalement la présence d’une insuffisance viscérale sous-jacente, l’âge élevé du patient, ou l’usage concomitant de certains médicaments comme les inhibiteurs de l’anhydrase carbonique.19
Conclusion
Les connaissances actuelles qui s’offrent au sujet de la physiologie fondamentale de l’insuline ont permis d’alléger la compréhension des mécanismes qui font que l’organisme ne soit pas parfaitement sensible à cette hormone chez certaines personnes. Même si on reste loin d’avoir totalement désenterré le trésor de savoir concernant ce sujet, le peu qu’on a entre nos mains, de nos jours, nous raccorde assez de lumières et de capacités d’avancer sur le volet thérapeutique afin de permettre un bien-être convenable et optimal aux sujets qui souffrent de la résistance à l’insuline.
Références
1. Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Na-ture. 2001 Dec 13;414(6865):782-7.
2. Santoleri D, Titchenell PM. Resolving the Paradox of Hepatic Insulin Resistance. Cell Mol Gas-troenterol Hepatol. 2019;7(2):447-456.
3.Patil PD, Mahajan UB, Patil KR, Chaudhari S, Patil CR, Agrawal YO, Ojha S, Goyal SN. Past and current perspective on new therapeutic targets for Type-II diabetes. Drug Des Devel Ther. 2017 May 22;11:1567-1583.
4. Rohner-Jeanrenaud F, Nogueiras R. Endocrine control of energy homeostasis. Mol Cell Endo-crinol. 2015 Dec 15;418 Pt 1:1-2.
5. Tokarz VL, MacDonald PE, Klip A. The cell biology of systemic insulin function. J Cell Biol. 2018 Jul 2;217(7):2273-2289.
6. Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011 Jul 6;14(1):9-19.
7. Ramnanan CJ, Edgerton DS, Kraft G, Cherrington AD. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab. 2011 Oct;13 Suppl 1(Suppl 1):118-25.
8. Artner I, Stein R. Transcriptional regulation of insulin gene expression. In : Pancreatic Beta Cell in Health and Disease. Springer, Tokyo 2008.pp : 13–30.
9. Kubota N, Kubota T, Kajiwara E, Iwamura T, Kumagai H, Watanabe T, Inoue M, Takamoto I, Sasako T, Kumagai K, Kohjima M, Nakamuta M, Moroi M, Sugi K, Noda T, Terauchi Y, Ueki K, Ka-dowaki T. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat Commun. 2016 Oct 6;7:12977.
10. Farese RV, Sajan MP, Standaert ML. Insulin-sensitive protein kinases (atypical protein kinase C and protein kinase B/Akt): actions and defects in obesity and type II diabetes. Exp Biol Med (Maywood). 2005 Oct;230(9):593-605.
11. Gross DN, Wan M, Birnbaum MJ. The role of FOXO in the regulation of metabolism. Curr Diab Rep. 2009 Jun;9(3):208-14.
12. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of choles-terol and fatty acid synthesis in the liver. J Clin Invest. 2002 May;109(9):1125-31.
13. Beale EG. Insulin signaling and insulin resistance. J Investig Med. 2013;61(1):11-14.
14. Stolerman ES, Florez JC. Genomics of type 2 diabetes mellitus: implications for the clinician. Nat Rev Endocrinol. 2009 Aug;5(8):429-36.
15. Ahima RS, Lazar MA. Adipokines and the pe-ripheral and neural control of energy balance. Mol Endocrinol. 2008;22:1023–31.
16. Zac-Varghese S, Tan T, Bloom SR. Hormonal interactions between gut and brain. Discov Med. 2010 Dec;10(55):543-52.
17. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219-46.
18. Petrie JR, Pearson ER, Sutherland C. Implications of genome wide association studies for the understanding of type 2 diabetes pathophysiology. Biochem Pharmacol. 2011 Feb 15;81(4):471-7.
19. Pfeiffer AF, Klein HH. The treatment of type 2 diabetes. Dtsch Arztebl Int. 2014 Jan 31;111(5):69-81; quiz 82.
20. Kelley DE, Wing R, Buonocore C, Sturis J, Polonsky K, Fitzsimmons M. Relative effects of ca-lorie restriction and weight loss in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1993 Nov;77(5):1287-93.
21. American Diabetes Association. Standards of medical care in diabetes- 2011. Diabetes Care. 2011;34 Suppl 1(Suppl 1):S11-S61.