
Néoplasies Endocriniennes Multiples – Vue d’Ensemble
Yanis AFIR & Rafik KORISSI
Les néoplasies endocriniennes multiples sont un groupe de pathologies génétiques relativement rares, dont la pathogénie est complexe et le traitement long et difficile. Cet article se propose de donner un aperçu des principaux syndromes néoplasiques endocriniens multiples en mettant en lumière leurs particularités cliniques, les mécanismes moléculaires entrant en jeu et les grandes lignes de la prise en charge et du suivi.
Définition/généralités
Les néoplasies endocriniennes multiples (NEM) sont un groupe de syndromes héréditaires de transmission autosomique dominante caractérisé par le développement de tumeurs principalement endocrines. Ces tumeurs peuvent être bénignes ou malignes, fonctionnelles ou non. Des tumeurs non endocrines peuvent également se voir.
Les premières descriptions de syndromes pluri-endocriniens familiaux semblent remonter au début des années 1900 avec les descriptions d’Erdheim,2 ensuite, à mesure que des études cliniques et génétiques étaient conduites, différentes entités furent individualisées.
Aujourd’hui on dénombre 4 types de NEM, chacun avec ses particularités cliniques, génétiques et thérapeutiques :
- NEM1 : c’est le type le plus fréquent, elle est caractérisée principalement par la triade : tumeur des glandes parathyroïdes, tumeur entéro-pancréatique et tumeur de l’antéhypophyse ;
- NEM2 : divisée en 2 sous-groupes
- NEM2A : caractérisée principalement par la triade : tumeur des glandes parathyroïdes, tumeur médullaire de la thyroïde et phéochromocytome ; et
- NEM2B (parfois appelée NEM3) : caractérisée par l’association de tumeur médullaire de la thyroïde et de phéochromocytome. À l’inverse de la NEM2A les parathyroïdes sont rarement touchées. Une multitude d’autres anomalies extra-endocriniennes sont généralement présentes et caractérisent également ce syndrome, elles seront décrites ultérieurement.
- NEM4 : autrefois assimilée à la NEM1 mais aujourd’hui totalement individualisée car le substratum génétique est différent.
Nous n’aborderons pas dans ce texte les autres syndromes endocriniens génétiques tels que le complexe de Carney, la maladie de von Hippel-Lindau ou le syndrome de McCune-Albright.
Le diagnostic de ces syndromes est théoriquement simple, la majorité des patients se présentant dans un contexte de polyendocrinopathie et/ou avec un contexte familial évocateur. Néanmoins, en pratique, la question s’avère beaucoup plus ardue. D’une part, le tableau clinique est souvent incomplet, les tumeurs n’apparaissant pas au même moment et les formes asymptomatiques étant fréquentes ; d’autre part, le contexte familial peut manquer du fait d’un décès précoce d’un parent, d’une apparition tardive des symptômes ou, souvent, l’apparition d’une mutation de novo.4
Le tout est d’y penser et de les rechercher systématiquement avec un interrogatoire précis, un examen clinique complet et éventuellement des examens complémentaires adéquats.
Une fois le diagnostic suspecté, une étude génétique pour le malade et l’ensemble de la famille est réalisée. Les principales mutations en cause sont aujourd’hui identifiées pour chacun des syndromes. La NEM1 résulte d’une mutation inactivatrice d’un gène suppresseur de tumeur appelé Ménine situé sur le chromosome 11. NEM2 quant à elle résulte d’une mutation activatrice sur le gène prooncogène RET sur le chromosome 10. La question est plus complexe concernant la NEM4 et sera abordée à la fin de ce texte.
À noter qu’il existe un parallélisme clinico-génétique important pour la NEM2 mais très faible pour la NEM1. Ce point sera détaillé par la suite.
Enfin, malgré les avancées des études moléculaires et génétiques, de multiples questions restent non résolues3 : pourquoi certaines glandes sont touchées et pas d’autres ? Pourquoi certaines tumeurs sont bénignes et d’autres malignes ? Quel est le rôle de l’hyperplasie dans le processus physiopathologique ? Quelles molécules peuvent être prises pour cibles dans le cadre d’un traitement étiologique ?
NEM1
La néoplasie endocrinienne multiple de type 1, aussi appelée syndrome de Wermer,7 fut la première des NEM à être individualisée. Comme indiqué précédemment, elle implique principalement 3 glandes : la parathyroïde, le pancréas endocrine avec le tube digestif, et l’antéhypophyse.
Le diagnostic peut être fait par l’un des 3 critères suivants :
• Critère clinique : la présence de 2 ou plus tumeurs du spectre NEM1 chez un même individu.
• Critère familial : la présence d’une tumeur du spectre NEM1 chez une personne dont un parent du premier degré est diagnostiqué NEM1.
• Critère génétique : l’identification d’une mutation du gène NEM1.
La prévalence de la maladie est variable dans le monde, en moyenne entre 2 et 20 cas par 100.000 habitants, touchant de manière égale les deux sexes.3 Plusieurs cas ont été déclarés en Algérie mais à l’heure actuelle on ne connait pas les chiffres exacts.
La transmission est autosomique dominante avec une pénétrance maximale (> 90%) aux environs de 40 ans. L’âge de début de la symptomatologie varie grandement (de 5 ans à plus de de 80 ans !3).
Enfin, 10% des cas de NEM1 résultent d’une mutation de novo. Ces cas sont dénommés NEM1 sporadiques.
Caractéristiques cliniques :
1- Hyperparathyroïdie : c’est l’anomalie la plus fréquente de la NEM1, présente chez environ 90% des patients. C’est aussi la première à apparaître dans la plupart des cas. Néanmoins, seuls 1 à 3% des hyperparathyroïdies sont dus à la NEM1, la majorité des cas étant sporadiques.
Il existe des différences notables entre l’hyperparathyroïdie sporadique et celle due à la NEM1.2 Tout d’abord, la forme génétique survient plus précocement que son homologue (25 ans vs 55 ans) et atteint de manière égale les deux sexes (alors que la forme sporadique est de prédominance féminine). Ensuite, elle se manifeste le plus souvent par une hyperplasie qui atteint toutes les glandes parathyroïdes, impliquant donc une chirurgie plus lourde et un plus grand risque d’hypoparathyroïdie post-chirurgicale. Enfin, le risque de cancérisation dans la forme génétique est quasi-nul.
2- Tumeurs entéro-pancréatiques : deuxième atteinte la plus fréquente, touchant environ 50% des patients. Néanmoins certains auteurs estiment que ce chiffre est nettement sous-estimé puisque des études autopsiques ont retrouvé des tumeurs pancréatiques cliniquement silencieuses chez la quasi- totalité des patients NEM1 de plus 40 ans.2
Parmi ces tumeurs, le gastrinome est le plus fréquemment retrouvé, il est responsable du syndrome de Zollinger-Ellison. Il est intéressant de mentionner que le quart de tous les cas de gastrinomes sont dus à la NEM1. Dans ce cadre, il survient plus précocement que dans les cas sporadiques (en moyenne 10 ans avant). Il est le plus souvent multiple, petit, multifocal et situé fréquemment dans le duodénum. Ce point est crucial pour la prise en charge thérapeutique.
La deuxième tumeur entéro-pancréatique la plus fréquemment retrouvée est l’insulinome (20% des patients). Ce dernier survient fréquemment chez les patients avant 40 ans (contrairement à son homologue sporadique) et des métastases sont retrouvées dans 50% des cas (versus 10% pour la forme sporadique).
Le reste des tumeurs entéro-pancréatiques inclut les tumeurs productrices de glucagon, de VIP et des tumeurs non fonctionnelles. Ces tumeurs, autrefois négligées, suscitent actuellement un engouement croissant à mesure que les études révèlent que leurs fréquences sont nettement sous-estimées et qu’elles seraient des éléments pronostiques majeurs.3
3- Tumeurs de l’antéhypophyse : elles touchent environ le tiers des patients et présentent la même distribution de fréquence que les tumeurs sporadiques (dans l’ordre : adénome à prolactine, à GH, à FSH/LH, à ACTH, à TSH). De ce fait, le prolactinome est la 3ème tumeur la plus fréquemment retrouvée chez les NEM1 après la tumeur parathyroïde et le gastrinome.
Comme leurs homologues sporadiques, les adénomes hypophysaires sont rarement malins. Cependant, ils auraient tendance à être plus volumineux et polyclonaux. Ils répondent également moins bien au traitement médical.
4- Autres signes classiques : de multiples manifestations cliniques ont été rapportées plus ou moins fréquemment dans la littérature. Nous en citons quelquesunes de manière non exhaustive.
Les tumeurs carcinoïdes sont plus fréquentes en présence d’une NEM1. Elles naissent quasi exclusivement des tissus dérivant de l’intestin primitif antérieur, à savoir l’estomac, les bronches et le thymus. Pour des raisons inconnues, les tumeurs carcinoïdes du thymus touchent plus fréquemment les hommes et celles des bronches plus fréquemment les femmes.
Des formations cutanées variées tels que des lipomes sous-cutanés, des collagénomes ou des angiofibromes (particulièrement au niveau de la lèvre supérieure) se voient chez un nombre important de patients. Bien que n’ayant aucune importance clinique, leur présence pourrait renforcer la présomption de diagnostic et conforter le médecin dans sa décision de dépistage de la NEM1.
Les pathologies thyroïdiennes pourraient être plus fréquentes chez les NEM1, même si à ce jour aucun lien n’a été démontré. Des tumeurs variées telles que des léiomyomes et des méningiomes ont été décrites.
Enfin, des cas de phéochromocytomes, normalement caractéristique de la NEM2, ont été décrits chez quelques patients.
5- Nouvelles données : hormis les signes classiques de la maladie, de nouvelles études pointent du doigt des manifestations autrefois inconnues et/ou sous-estimées. Parmi elles nous en évoquons 2 : les tumeurs de la corticosurrénale et le cancer du sein.
Les anciennes études attribuaient une fréquence d’environ 5% aux différentes tumeurs corticosurrénales mais les études les plus récentes montrent que ce chiffre est nettement sous-estimé et qu’il serait en réalité proche de 70%10! La raison en est peut-être que la plupart de ces lésions sont non fonctionnelles. Lorsqu’elles sont cliniquement parlantes, elles se manifestent le plus souvent par un syndrome de Cushing ou un hyperaldostéronisme primaire. Les patients NEM1 seraient également plus à risque de développer un corticosurrénalome. La corrélation entre la NEM1 et le cancer du sein a été confirmée par de récentes études en Hollande. Le risque relatif est estimé à environ 2.8. Pour l’heure, le mécanisme proposé est que la protéine Ménine (voir plus bas) serait un co-activateur du récepteur alpha des oestrogènes. Les patients NEM1 résisteraient également plus à l’hormonothérapie.10
Substratum génétique et moléculaire :
En 1988, l’équipe menée par Larsson a identifié la région responsable de la NEM1 au niveau du bras long du chromosome 11 (11q13). Une décennie plus tard, le gène fut identifié et désigné sous le nom de « Ménine ».6
Il fut observé de fréquentes pertes d’ADN à ce niveau. De ce fait, il fut proposé que le gène Ménine agit comme suppresseur de tumeur et obéissait dans son fonctionnement à l’hypothèse de Knudson :
L’allèle défectif reste silencieux lorsque le second allèle est normal ; ceci est le cas pour la plupart des cellules. On dit que les cellules gardent leur état d’hétérozygote et sont de ce fait protégées de l’allèle défectueux par l’allèle normal.
Dans certaines cellules, il y a perte de l’état hétérozygote lorsqu’une seconde mutation survient sur l’allèle resté normal (le plus souvent une délétion), les cellules commencent à proliférer de manière anormale.
Ces mutations sur le second allèle surviendraient de manière très fréquente et expliqueraient en partie la transmission autosomique dominante de la pathologie.1
Bien que ce modèle s’applique à la plupart des patients, dans moins de 10% des cas, aucune perte de l’état hétérozygote n’est retrouvée, parfois même il n’y a aucune mutation sur aucun des allèles. Il y aurait donc d’autres régions impliquées dans la régulation et le contrôle de ce gène à élucider par de futures recherches.
Revenons au gène Ménine. Il code pour une protéine appelée… Ménine aussi. Il s’agit d’une protéine à 610 acides aminés dont les fonctions sont multiples et non encore totalement comprises. Plus de 1300 mutations possibles ont été identifiées sur tout le long de la protéine. Néanmoins, la corrélation entre les mutations et la présentation clinique est très faible et il est, pour l’heure, impossible de prédire le phénotype des patients en se basant sur l’étude génétique seule.
Ménine ne possède pas de fonction enzymatique claire ou une séquence structurale typique permettant de deviner son rôle. Ses fonctions ont donc été largement déduites de ses interactions avec d’autres protéines.9 Aujourd’hui, au moins 4 fonctions ont été identifiées : régulation de la transcription, stabilité du génome, régulation de la division cellulaire et enfin régulation épigénétique.3
En ce qui concerne la transcription, Ménine inhibe plusieurs facteurs et voies de transcription tels que JunD, la famille NF-kB, la famille Smad… etc. Elle se lierait aussi directement à l’ADN pour inhiber l’effet des Insulin-like Growth Factor Binding Protein 2 (IGFBP2) ou activer des caspases (processus d’apoptose).
Pour son rôle dans la stabilité du génome, l’hypothèse a été émise suite à l’identification de multiples interactions entre Ménine et différentes protéines de la réplication et de la correction génomique.
Enfin, au niveau épigénétique, Ménine agirait en régulant la méthylation et l’acétylation des histones. Particulièrement, il a été démontré que Ménine interagissait avec les protéines du complexe MLL (Mixed-Lineage Leukemia). Pour rappel, certaines translocations du gène MLL1 lui permettent d’activer l’expression du gène HOX qui est en cause dans les leucémies. Il a été observé que l’interaction de Ménine avec MLL
était essentielle dans le processus oncogénique. De ce fait, des inhibiteurs de l’interaction Ménine-MLL ont été développés et les résultats des études in vitro et sur animal sont prometteuses et donneront peut-être lieu à des essais cliniques.9
Prise en charge :
Le traitement des tumeurs de la NEM1 est plus complexe que celui des tumeurs sporadiques. En effet, les tumeurs associées à la NEM1 sont généralement multiples, plus larges, plus infiltrantes et résistantes aux traitements médicamenteux.
Les métastases occultes sont également plus fréquentes. Pour l’hyperparathyroïdie, le traitement est généralement chirurgical avec l’ablation de toutes les glandes parathyroïdes accessibles. Néanmoins, il est très difficile de toutes les individualiser, surtout que les glandes surnuméraires orthotopiques ou ectopiques sont fréquentes.
Une thymectomie prophylactique est généralement réalisée pour enlever d’éventuels tissus parathyroïdiens intra-thymiques et d’éventuelles tumeurs carcinoïdes thymiques.
La particularité de l’hyperparathyroïdie du NEM1 est le fort risque de persistance ou de récidive après traitement.
La persistance de l’hyperparathyroïdie malgré un traitement chirurgical survient dans environ 12% des cas et elle serait probablement due à des glandes parathyroïdes surnuméraires ou ectopiques.
La récidive quant à elle est beaucoup plus fréquente, surtout lorsque l’on n’enlève pas toutes les glandes, survenant dans près de 45% des cas. En effet même si la glande touchée a été enlevée, le stimulus mitogénique persiste toujours sur les glandes restantes et elles peuvent tuméfier à tout moment.
Pour les cas récidivants, une chirurgie complémentaire donne en général de bons résultats.
Pour les gastrinomes, la chirurgie est très difficile, voire quasiimpossible, étant donnée la nature petite, multiple et ectopique des tumeurs. Certains centres ont proposé de réaliser une DPC (Duodéno-Pancréatectomie Céphalique) d’emblée à tous les patients, mais le rapport bénéfice/risque serait loin d’être favorable.
En Algérie, cette procédure est réservée aux patients ayant une tumeur volumineuse nettement identifiée dont le diagnostic est certain et chez qui la chirurgie est réalisable.
Pour l’heure, le traitement médicamenteux à base d’inhibiteurs de la pompe à proton (IPP) reste le gold standard.
Un point important à considérer est qu’on ne peut pas évaluer la sévérité du syndrome de Zollinger-Ellison avant d’avoir au préalable traité l’hyperparathyroïdie. En effet, le calcium est un stimulateur de la sécrétion gastrique et l’hypercalcémie aggrave les symptômes dus à l’hyperacidité gastrique. D’ailleurs, il est fréquent de constater une nette diminution des doses d’IPP nécessaires pour contrôler l’hyperacidité après chirurgie des parathyroïdes.
Les insulinomes, contrairement aux gastrinomes, se localisent souvent au niveau du pancréas et un traitement chirurgical peut être envisagé. Même chose pour les glucagonomes et les VIPomes où le traitement chirurgical est recommandé.
Les indications de traitement des tumeurs non fonctionnelles sont plus complexes et plus controversées. Retenons juste que plus la tumeur est de grande taille (généralement > 2 cm) plus elle a de potentiel de malignité et plus le traitement chirurgical trouve son indication.
Concernant les tumeurs antéhypophysaires, le traitement préconisé est généralement le même que pour les tumeurs sporadiques, à savoir un traitement par agonistes dopaminergiques pour les prolactinomes et un traitement chirurgical pour les autres lignées. Rappelons tout de même que la prise en charge est plus difficile, les échecs au traitement médicamenteux sont plus fréquents et les tumeurs plus difficilement résécables.
La meilleure compréhension de ces pathologies et l’introduction de traitements adéquats ont bouleversé le pronostic de la NEM1.
La littérature rapporte une mortalité à plus de 50% à 50 ans en l’absence de traitement. Auparavant, la principale cause de décès était les différentes complications du syndrome de Zollinger- Ellison. Mais l’épidémiologie a été complètement bouleversée par l’introduction des IPP. Aujourd’hui, les principales causes de mortalités sont les tumeurs pancréatiques non sécrétantes et les tumeurs carcinoïdes (thymiques particulièrement).
En l’absence de ces tumeurs, l’espérance de vie est d’environ 60 ans. En leur présence, l’espérance de vie tombe à un peu plus de 40 ans. Pour les patients dont le décès n’est pas directement lié à une tumeur de la maladie, les complications cardiovasculaires semblent être la principale cause de décès. Les patients NEM1 seraient à risque cardiovasculaire possiblement àcause de l’hyperinsulinisme et des hypoglycémies.
Ceci étant dit, l’absence de données ne permet pas de statuer sur le pronostic en Algérie, néanmoins, est constatée une nette diminution de l’espérance de vie, avec un pronostic plus péjoratif que pour les autres types de NEM.
Dépistage et suivi :
Lorsqu’un cas de NEM1 est suspecté ou confirmé. Le patient ainsi que ses parents du premier degré doivent bénéficier d’un dépistage adéquat et d’un suivi régulier.
Tout d’abord, une étude génétique doit être entreprise. Néanmoins comme nous l’avons vu précédemment, la corrélation clinico-génétique est très faible et les résultats de l’étude génétique ne permettent pas de conditionner les méthodes de dépistage futures.
Le suivi doit commencer le plus tôt possible, car les manifestations peuvent commencer très tôt et rester longtemps asymptomatiques. Il se basera sur un interrogatoire et un examen clinique précis et toute une batterie d’examens biologiques et radiologiques spécifiques à des intervalles plus ou moins rapprochés.
Inutile de détailler les modalités du suivi, disons juste qu’un suivi rapproché et intensifié doit être poursuivi au moins jusqu’à 40-45 ans, âge où la pénétrance de la maladie est supérieure à 90%. Si à cet âge-là aucune manifestation n’est apparue, le suivi peut être allégé par la suite mais poursuivi tout de même.
Enfin, il est légitime de se demander si le dépistage systématique de la NEM1 doit être entrepris devant un patient présentant une tumeur appartenant au spectre mais de manière isolée.
Comme il a été dit plus haut, la plupart des cas d’hyperparathyroïdie et de tumeurs antéhypophysaires sont sporadiques ; un dépistage systématique de la NEM1 dans ces cas ne semble pas justifié. Par contre, une proportion considérable des gastrinomes et des tumeurs carcinoïdes thymiques est due à la NEM1, un dépistage semble intéressant dans ces cas.
Le dépistage est également recommandé devant l’apparition d’une symptomatologie à un âge précoce (généralement < 30 ans).
Reproduit à partir de Gardner, D. G., Shoback, D. M., & Greenspan, F. S. (2017). Greenspan’s basic & clinical endocrinology. New York: McGraw-Hill Medical.
NEM2
La néoplasie endocrinienne multiple de type 2 a été distinguée du type 1 vers la fin des années 60.2 Elle regroupe 2 syndromes indépendants :
• NEM2A : aussi appelée syndrome de Sipple,8 elle regroupe la triade : carcinome médullaire de la thyroïde, phéochromocytome et hyperparathyroïdie. Elle représente 95% des NEM2 et peut être elle-même divisée en 4 variantes (détaillées plus loin).
• NEM2B : aussi appelée syndrome de Gorlin par certains auteurs, elle associe carcinome médullaire de la thyroïde et phéochromocytome avec une variété d’autres manifestations ; l’hyperparathyroïdie est beaucoup plus rarement retrouvée.
La prévalence est d’environ 1 à 10 cas par 100.000 habitants. La pénétrance est plus faible que dans la NEM1, approximativement 70% à 70 ans. Comme
pour la NEM1, nous n’avons pas de chiffres exacts de la prévalence en Algérie.
Caractéristiques cliniques :
1- Caractéristiques cliniques communes : le carcinome médullaire de la thyroïde (CMT) est la manifestation la plus fréquente de la NEM2 (environ 90% des cas) et est souvent la première à apparaître. Aussi, plus de 25% de tous les CMT sont dus à la NEM2.
Le phéochromocytome survient chez environ 50% des patients, le plus souvent après l’apparition du CMT mais parfois avant (point très important), surtout en cas de NEM2B.
Il est le plus souvent localisé au niveau de la médullosurrénale, fréquemment bilatéral et rarement malin. Un phéochromocytome unilatéral se bilatéralise souvent au bout de quelques années.
Un point intéressant, les phéochromocytomes dus à la NEM2 ont souvent une sécrétion d’adrénaline prédominante par rapport à la noradrénaline, contrairement aux formes sporadiques où dues à d’autres pathologies génétiques (comme le syndrome de von Hippel-Lindau) où la sécrétion prédomine sur la noradrénaline.
Le point crucial concernant le phéochromocytome est son dépistage systématique devant la découverte de tout CMT, surtout avant un traitement chirurgical. En effet, opérer un patient qui a un phéochromocytome non traité peut entrainer des complications potentiellement mortelles.
L’hyperparathyroïdie peut se voir dans environ 25% des cas, la majorité étant des NEM2A, beaucoup plus rarement NEM2B. Elle est moins agressive que lorsqu’elle est due à la NEM1 et répond beaucoup mieux au traitement.
Tiré de Castinetti F, Moley J, Mulligan L, Waguespack SG. A comprehensive review on MEN2B. Endocr Relat Cancer. 2018;25(2):T29–T39.
2- Variantes cliniques de la NEM2A : on distingue en réalité 4 types de NEM2A : d’abord la NEM2A classique (décrite plus haut), avec 3 autres variantes cliniques : le carcinome médullaire de la thyroïde familial, la NEM2A associée à une maladie de Hirschsprung et la NEM2A associée au Lichen amyloïde.
Auparavant, les chercheurs considérèrent le carcinome médullaire de la thyroïde familial comme une maladie à part. Néanmoins, comme le CMT est souvent la première manifestation de la NEM2, on ne savait pas quels patients avaient réellement une forme familiale de CMT isolée et lesquels étaient des NEM2 qui pouvaient développer un phéochromocytome. Étant donné le danger de cette seconde éventualité, il a été proposé une définition très stricte du CMT familial : une famille comportant au moins 10 membres ayant un CMT ou avec de multiples porteurs ou patients atteints après l’âge de 50 ans, en excluant la présence de phéochromocytome et d’hyperparathyroïdie.
À ce jour seules 3 familles dans le monde rentrent dans ces critères.2
Maintenant, la plupart des chercheurs, confortés par les résultats des études génétiques, considèrent le CMT familial comme une forme atténuée de NEM2 et certains formulent même l’hypothèse que les autres manifestations de la maladie pourraient apparaître à condition que les patients vivent assez longtemps.
Dans la deuxième variante de la NEM2A, elle est associée à la maladie de Hirschsprung (mégacôlon congénital) due à une aganglionose entérique congénitale.
Enfin, le Lichen amyloïde est caractérisé par des dépôts cutanés kératosiques. Ils sont précédés par une longue phase de prurit cutané intense. Les lésions de grattage induisent à la longue des indurations cutanées et des dépôts de kératine.
Contrairement à la NEM1, chaque variante de NEM2 est associée à une mutation génétique particulière.
Il est possible de prédire le phénotype du patient en se basant sur l’étude génétique.
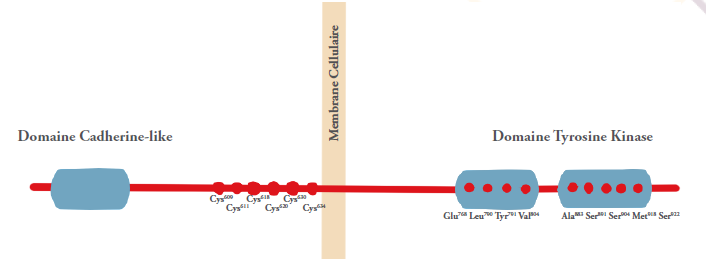
3- Autres signes cliniques de la NEM2B : le patient NEM2B présente souvent un faciès caractéristique avec l’association d’anomalies morphologiques générales comme une apparence marfanoïde, un thorax en entonnoir, une scoliose… etc. avec des anomalies faciales typiques : des paupières épaisses et écartées, un ptosis et de larges nerfs cornéens… etc. (Figure 2).
Substratum génétique et moléculaire :
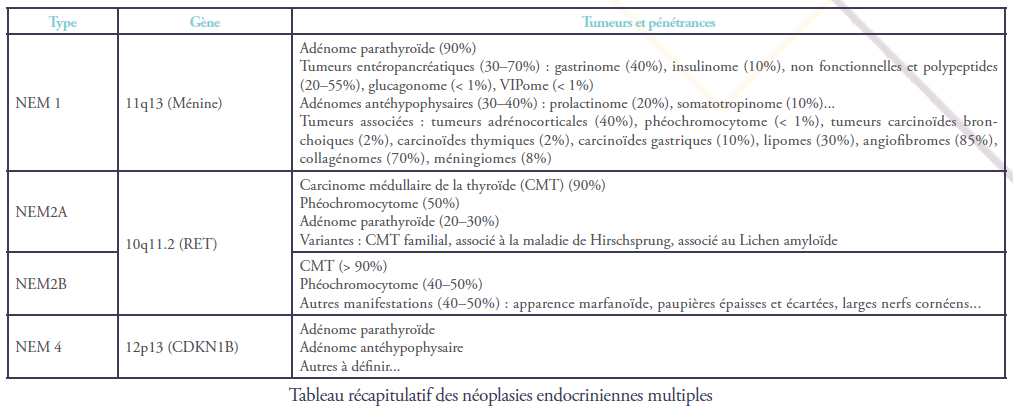
Le gène responsable de la NEM2 est localisé au niveau du chromosome 10 (10q11.2) et est appelé RET. RET, pour REarranged during Transfection (appelé comme ça car il a été retrouvé recombiné lors de la transfection d’une cellule… bref ), est un gène qui code pour une protéine appelée… RET (ahah !).
Il s’agit d’une protéine membranaire à 3 domaines (extra-cellulaire, transmembranaire et intra-cellulaire) à activité tyrosine kinase (Figure 1). Elle joue un rôle essentiel dans la régulation de la différenciation, la prolifération, la migration et la survie des cellules.
La protéine RET comporte un composant structural particulier avec une série de résidus cystéines successifs qui jouerait un rôle inhibiteur et régulateur de l’activité de la protéine dans les cellules normales. C’est justement au niveau de ces résidus que la plupart des mutations
surviennent.
Les mutations sont différentes dans les NEM2A et NEM2B et le mécanisme de l’hyperactivité de la protéine RET est également différent dans chaque cas.
Dans lq NEM2A, ce sont surtout les régions extracellulaire et transmembranaire qui sont touchées par les mutations, induisant une perte d’un résidu cystéine.
En conséquence, les protéines RET peuvent s’auto-activer indépendamment de la présence de ligands. La mutation la plus fréquente dans ce cas touche le codon 634 et substitue une arginine à la cystéine (C634R).
Dans la NEM2B, la mutation touche le plus souvent la partie intra-cellulaire de la protéine. En conséquence, la spécificité de la protéine envers ses effecteurs est altérée, et elle devient capable d’activer d’autres voies intracellulaires. La mutation la plus fréquente dans ce cas touche le codon 918 et substitue une thréonine à une méthionine (M918T).
Il est intéressant de dire que 50% des NEM2B sont dus à une mutation de novo et que celle-ci survient quasi exclusivement sur l’allèle paternel, pour
des raisons encore inconnues.
Concernant les variantes de NEM2A. Il a été observé que les patients atteints de CMT familial avaient des mutations similaires à ceux ayant un phénotype NEM2A classique, ce qui voudrait dire qu’il existe des mécanismes additionnels qui réguleraient l’expression du gène RET.
La maladie de Hirschsprung implique des mutations dans les codons 609, 611, 618, et 620, et le Lichen amyloïde implique le codon 634.
Un point très intéressant. On a dit que les NEM2 sont dues à des mutations du gène RET qui induisent un gain de fonction anormal ; ceci n’est pas tout à fait vrai pour la maladie de Hirschsprung. En effet, le gène RET est essentiel au développement embryonnaire de plusieurs structures, dont les ganglions myentériques. C’est donc une inactivation du gène RET qui provoque la maladie de Hirschsprung et non son hyper-activation.
Donc, dans les formes de NEM2A associée à la maladie de Hirschsprung, il existe à la fois une hyperactivation et une inhibition du RET en fonction des cellules !
Prise en charge :
Le traitement du CMT est classique : chirurgie avec large curage ganglionnaire (cancer très lymphophile) en utilisant le taux de calcitonine pour le suivi postopératoire.
Le plus souvent les patients NEM2B sont opérés plus précocement que les NEM2A car les CMT y sont souvent plus agressifs.
La particularité ici, rappelons-le, est la nécessité de vérifier l’absence de phéochromocytome et de le traiter si présent avant d’opérer le patient.
Le traitement du phéochromocytome est similaire aux cas sporadiques. Les alpha-bloquants sont utilisés comme traitement symptomatique en attendant la chirurgie. Cette dernière consiste le plus souvent en une adrénalectomie unilatérale avec surveillance de la glande controlatérale. En cas de phéochromocytome touchant les deux glandes, le traitement sera le plus conservateur possible.
De nouvelles thérapeutiques prometteuses sont en cours d’étude. Il s’agit des inhibiteurs de l’activité tyrosine kinase de RET, vandetanib et cabozantinib. Si ceux-ci sont approuvés pour le traitement, ils s’avéreront particulièrement intéressants pour les cas avancés ou inopérables de CMT et ce sera la première fois qu’on proposera un traitement « étiologique » à la NEM2.
Le pronostic de la maladie a été largement amélioré par le suivi et le traitement adéquat. La principale cause de mortalité demeure le CMT.12
Dépistage et suivi :
Tout comme pour la NEM1, dès qu’un cas de NEM2 est suspecté ou confirmé, le patient ainsi que ses parents du premier degré doivent bénéficier d’un dépistage adéquat et d’un suivi régulier. Il est généralement recommandé de rechercher une NEM2 chez toute personne présentant un CMT. Il subsiste des controverses quant à l’attitude à adopter lorsqu’une mutation est identifiée chez un membre sain de la famille. Actuellement, des paramètres génétiques et biochimiques permettent d’évaluer le risque de développer un cancer et une thyroïdectomie prophylactique est proposée dans ces cas. Exception faite des patients NEM2B ou dans des formes particulièrement agressives de CMT familial où une thyroïdectomie
prophylactique est proposée à tous les patients.
NEM4
C’est la dernière-née des NEMs. Évidemment c’est la plus rare et la moins étudiée.15 Elle a été découverte en étudiant des cas de NEM1 chez qui l’on n’avait retrouvé aucune mutation du gène Ménine mais qui présentaient des mutations similaires dans un autre gène : CDKN1B.
Ce syndrome a été d’abord décrit chez les rats et fut appelé NEMX mais un premier cas humain a été rapporté en 2006 par l’équipe de Pellegata.15 Suite à cela, le syndrome NEM4 a été officiellement introduit lors de la 11ème conférence internationale des NEM en 2008.
Depuis, seuls 19 cas ont été identifiés dans le monde. La présentation clinique est très similaire à celle des NEM1, avec tout de même quelques différences notables.
L’hyperparathyroïdie est la manifestation la plus fréquente (80%). Elle ressemble plus aux cas sporadiques qu’à son homologue de NEM1. En effet elle survient à un âge plus tardif, touche de préférence les femmes et jusqu’à l’heure aucun cas de récidive n’a été rapporté.
Les tumeurs antéhypophysaires et entéro-pancréatiques semblent également moins agressives que dans les cas de NEM1. Il n’y a pour l’instant aucun cas décrit d’insulinome, de VIPome, de glucagonome ou de tumeur non sécrétante maligne.
Le reste des manifestations de la NEM1, notamment cutanées, est plus rarement retrouvé.
Enfin, la NEM4 aurait la particularité d’être impliquée dans l’insuffisance ovarienne primitive.
Concernant le substratum génétique, le gène impliqué est le CDKN1B, situé sur le chromosome 12 (12p13), qui code pour la protéine p27 qui régule le cycle cellulaire. La transmission est autosomique dominante (contrairement à la NEMX décrite chez les rats où la transmission est récessive).16
Même si les études n’en sont encore qu’à leurs débuts, on sait déjà que le gène n’obéit pas forcément au modèle de Knudson et qu’il existe des interactions bilatérales entre CDKN1B et Ménine.
Conclusion
Il apparaît à la fin de cet exposé long et ennuyeux (pour vous qui êtes arrivés à la fin par patience, passion ou compassion) que les néoplasies endocriniennes multiples représentent un problème sérieux et complexe, déjà sur le plan théorique. Nous ne pouvons donc qu’imaginer les difficultés surajoutées lors de la confrontation avec la réalité pratique. Il est donc plus que nécessaire de coordonner les efforts locaux en établissant par exemple un registre national des NEM et en désignant des référants nationaux sur le sujet afin de prendre en charge au mieux nos patients.
Références
1- Gardner, D. G., Shoback, D. M., & Greenspan, F. S. (2017). Greenspan’s basic & clinical endocrinology. New York: McGraw-Hill Medical.
2- In Melmed, S., In Polonsky, K. S., In Larsen, P. R., & In Kronenberg, H. (2016). Williams textbook of endocrinology.
3- Jameson, J. L., DeGroot, L. J., & De, K. D. M. (2016). Endocrinology: Adult and pediatric. Philadelphia: Saunders/Elsevier.
4- DynaMed Plus [Internet]. Ipswich (MA): EBSCO Information Services. 1995 – . Record No. T113809, Multiple endocrine neoplasia type 1; [updated 2018 Nov 30, cited 27/03/2019]
5- Rebecca Gorrigan, Maralyn Druce. Multiple endocrine neoplasia syndromes. BMJ Best Practice. Feb 2018.
6- Andrew Arnold. Multiple endocrine neoplasia type 1: Definition and genetics (2018). Jean E Mulder, MD (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
7- Cornelis J Lips, Douglas W Ball. Classification and genetics of multiple endocrine neoplasia type 2 (2018). Jean E Mulder, MD (Ed.), UpTo- Date. Waltham, MA: UpToDate Inc.
8- Cornelis J Lips, Douglas W Ball. Clinical manifestations and diagnosis of multiple endocrine neoplasia type 2 (2018). Jean E Mulder, MD (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
9- Cornelis J Lips, Douglas W Ball. Approach to therapy in multiple endocrine neoplasia type 2 (2018). Jean E Mulder, MD (Ed.), UpToDate. Waltham, MA: UpToDate Inc.
10- Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. 1954;16:363-371.
11- Sipple J. The association of pheochromocytoma with carcinoma of the thyroid gland. Am J Med. 1961;31:163-166.
12- Agarwal SK. The future: genetics advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer. 2017;24(10):T119.
13- van Leeuwaarde RS, de Laat JM, Pieterman CRC, Dreijerink K, Vriens MR, Valk GD. The future: medical advances in MEN1 therapeutic approaches and management strategies. Endocr Relat Cancer. 2017 Oct; 24(10):T179-T193.
14- Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN 1). J Clin Endocrinol Metab. 2012;97:2990-3011.
15- Joanna G, Kym W. Patient quality of life and prognosis in multiple endocrine neoplasia type 2. Endocrine-Related Cancer 2018. 25 T69– T77.
16- Castinetti F, Moley J, Mulligan L, Waguespack SG. A comprehensive review on MEN2B. Endocr Relat Cancer. 2018;25(2):T29–T39.
17- Asai N, et al. RET receptor signaling: dysfunction in thyroid cancer and Hirschsprung’s disease. Pathol. Int. 2006;56:164–172.
18- Alrezk R, Hannah-Shmouni F, Stratakis CA. MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer. 2017.
19- Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4) Mol Cell Endocrinol. 2014;386(1–2):2
20- Walls GV. Multiple endocrine neoplasia (MEN) syndromes. Semin Pediatr Surg. 2014;23(2):96–101.