La Maladie de Wilson
Nour El Houda MAHDI
La maladie de Wilson est une affection génétique qui résulte d’un déficit dans le transport hépatocellulaire du cuivre, conduisant à son accumulation toxique dans le foie et d’autres tissus. Elle est consécutive à des mutations du gène ATP7B et entraîne des manifestations cliniques très variables d’ordres hépatique, neurologique, hématologique et oculaire. Diagnostiquée sur un faisceau d’arguments, cette pathologie est l’une des rares maladies génétiques à pouvoir être traitée efficacement. Le traitement repose sur des chélateurs du cuivre associés à un régime pauvre en ce métal.
Introduction
La maladie de Wilson a été décrite pour la première fois en 1912 par Kinnear Wilson, en tant que maladie familiale caractérisée par un dysfonctionnement neurologique létal progressif avec cirrhose du foie et une anomalie cornéenne caractéristique : l’anneau de Kayser-Fleischer (KF). Dans cette maladie, l’excrétion hépatique inadéquate du cuivre entraîne son accumulation dans le foie, le cerveau, les reins et la cornée. La prévalence estimée dans la plupart des populations est de l’ordre de 1 cas sur 30 000 naissances.4
Physiologie du cuivre
En raison de ses propriétés redox, le cuivre est un métal essentiel, agissant comme un cofacteur polyvalent de nombreuses enzymes essentielles.1 Il s’agit essentiellement de la Lysyl Oxydase, impliquée dans la production de tissu conjonctif et la réticulation de l’élastine ; la Superoxyde Dismutase Cu/Zn, un piégeur (scavenger) de radicaux libres cytoplasmique ; la Cytochrome C Oxydase, qui fait partie intégrante de la phosphorylation oxydative mitochondriale ; la Tyrosinase, nécessaire à la production de pigments ; et enfin la β-dopamine Monooxygénase, impliquée dans la neurotransmission.4
Bien que l’organisme ait besoin du cuivre pour son bon fonctionnement, il utilise des quantités inférieures à celles qui sont apportées par l’alimentation. Une fois absorbé dans l’intestin, le cuivre se lie à l’albumine sérique et à divers acides aminés, notamment l’histidine, qui le distribuent vers différents tissus, dont principalement le foie.1,4 Le cuivre faiblement lié aux acides aminés est filtré par les reins et réabsorbé ultérieurement.
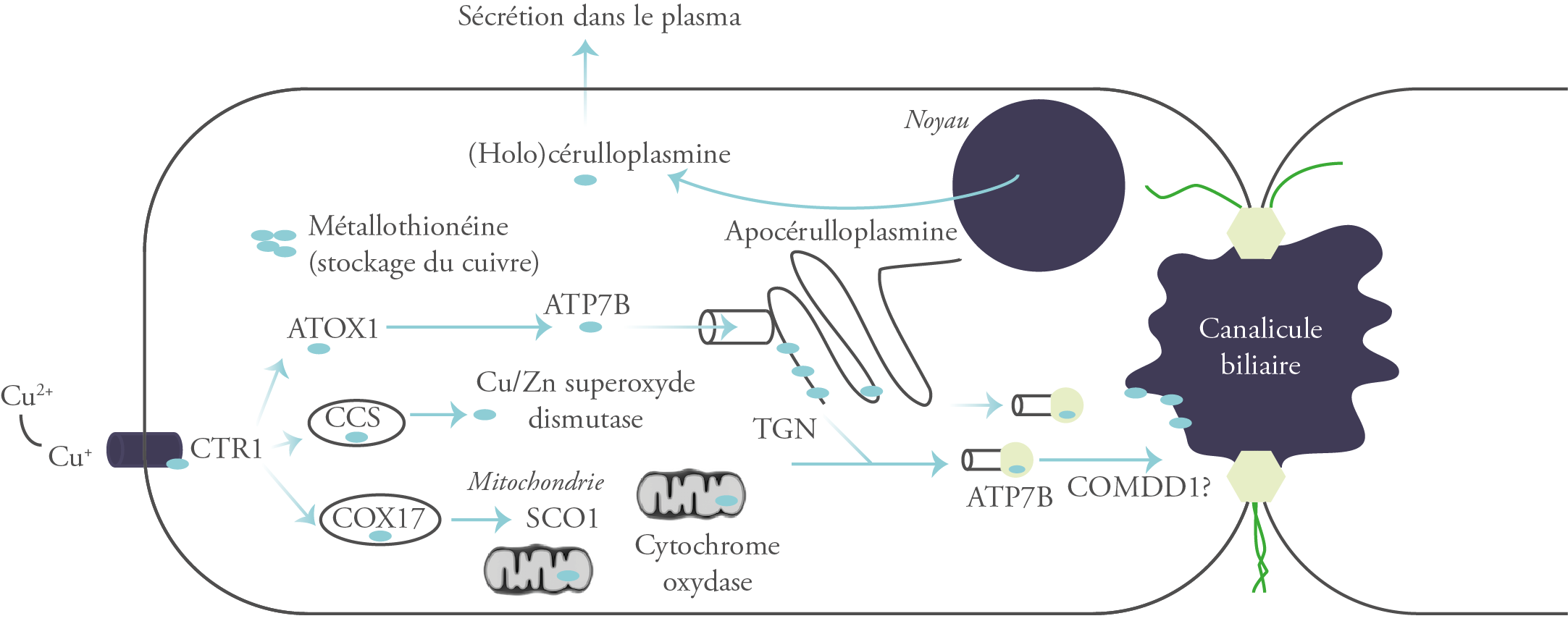
Au niveau du foie, le cuivre est internalisé dans les hépatocytes par le transporteur CTR1 (Human Copper Transporter 1). À l’intérieur de la cellule, il n’est jamais libre. Il se lie aux métallochaperones (ATOX1, SCO1/COX17 et CCS), des protéines de faible poids moléculaire dont chacune délivre ce métal à une protéine cible spécifique : l’ATP7B, Wilson-ATPase ; la Cytochrome C Oxydase ; et la Superoxyde Dismutase, respectivement.4
Lorsque les cellules hépatiques ou intestinales sont surchargées en cuivre, les métallothionéines, une classe de protéines riches en cystéine de faible poids moléculaire, sont activées et séquestrent le cuivre sous une forme non toxique. Le cuivre peut aussi former des complexes avec le glutathion intracellulaire.4
La voie prédominante de l’excrétion du cuivre est la voie hépatobiliaire. Les reins ne représentent que moins de 5% de l’excrétion totale, sauf si la capacité de réabsorption tubulaire rénale est dépassée, comme il se produit dans la maladie de Wilson.1
Pathogénèse de la maladie
Il existe deux maladies humaines impliquant le transport du cuivre qui sont dues à des adénosines triphosphatases de type P transportant des métaux (ATPases) : (1) la maladie de Menkes liée au chromosome X, qui est un défaut de transport du cuivre à partir de l’intestin dû à des mutations de l’ATP7A entraînant une carence en cuivre ; et (2) la maladie de Wilson, un trouble autosomique récessif dû à des mutations de l’ATP7B, qui entraîne au contraire une surcharge en cuivre.4
Le gène anormal dans la maladie de Wilson (ATP7B) se trouve sur le chromosome 13. Il a été cloné par une combinaison d’analyse de liaison conventionnelle et de cartographie physique de la région concernée du chromosome 13 (13q14) et, enfin, en se basant sur son homologie étendue avec le gène de la maladie de Menkes.1
Reproduite à partir de Sanyal, Arun J. Zakim and Boyer’s Hepatology. SAUNDERS, 2017. Fig 59-2, page 928.
Ce gène code pour l’ATP7B, ou Wilson-ATPase, une protéine transmembranaire de type P ATPase localisée préférentiellement dans le foie, mais aussi dans le cerveau. Elle a un domaine de liaison au cuivre composé de 6 unités de liaison en tandem, chacune présentant le motif CXXC, en plus d’une région ATPase en boucle et de 8 segments transmembranaires qui forment un pore.1
L’ATP7B présente 2 localisations subcellulaires : le réseau trans-Golgi et les vésicules cytoplasmiques.
Dans le réseau trans-Golgi, l’ATP7B assure la médiation du transport du cuivre pour son incorporation dans l’apocéruloplasmine pour produire la céruloplasmine, une α2-glycoprotéine qui contient 6 molécules de cuivre4 et qui possède une activité enzymatique similaire à la ferroxidase. À noter qu’environ 95% du cuivre du plasma est intégré au sein de la céruloplasmine.2
Dans les vésicules cytoplasmiques, le transport du cuivre médié par l’ATP7B séquestre l’excès de cuivre dans des vésicules pré-lysosomales, qui sont ensuite excrétées dans la bile par exocytose à travers la membrane canaliculaire apicale des hépatocytes et ultérieurement éliminées dans les selles.2, 4, 7
Les études in vitro utilisant diverses lignées cellulaires continues ont montré que l’emplacement intracellulaire de la Wilson-ATPase change avec la concentration de cuivre. Lorsque la concentration de cuivre intracellulaire est élevée, la Wilson-ATPase se redistribue du réseau trans-Golgi aux vésicules dans la région apicale de l’hépatocyte : à savoir, le voisinage de la membrane canaliculaire biliaire.1
Plus de 500 mutations de l’ATP7B ont été décrites, la H1069Q est la plus fréquente. Ces mutations altèrent le fonctionnement de la protéine ATP7B et donc l’excrétion biliaire du cuivre et son incorporation dans la céruloplasmine. Ceci entraîne une accumulation excessive de cuivre dans les hépatocytes. Ce cuivre est lié initialement à la métallothionéine. Lorsque la capacité tampon de cette dernière est dépassée, le cuivre est libéré dans les cellules, endommageant ainsi les hépatocytes.2
La céruloplasmine dépourvue de cuivre (apocérulloplasmine) a une demi-vie plasmatique brève et donc une concentration faible, ce qui contribue au diagnostic de la maladie.5
Conséquences de la surcharge en cuivre
Les mécanismes des lésions hépatocytaires dues au cuivre ne sont pas complètement élucidés. La nécrose et l’apoptose peuvent être rencontrées.2 La nécrose est principalement due à des lésions oxydatives liées à une production accrue de radicaux libres car le cuivre est un pro-oxydant et induit une capacité de réduction cellulaire altérée qui se traduit par de bas niveaux de glutathion réduit.2 Cette observation s’est appuyée sur la découverte d’un dysfonctionnement mitochondrial sévère7 associé à des lésions oxydantes accrues des lipides et de l’ADN.8
L’apoptose, quant à elle, est liée à la sécrétion induite par le cuivre de la sphingomyélinase à partir des leucocytes, ce qui libère la céramide, entraînant une mort hépatocytaire accrue et, enfin, une cirrhose du foie.
La céramide déclenche également l’apoptose des érythrocytes, par une exposition de la phosphatidylsérine à la surface des cellules qui déclenche leur phagocytose, avec pour conséquence une anémie hémolytique.9
Une teneur accrue en cuivre hépatique combinée à des dommages hépatocytaires entraîne la libération de cuivre dans le sang, et conduit à une augmentation de sa concentration sérique sous forme libre (cuivre non lié à la céruloplasmine), bien que les niveaux sériques totaux de cuivre puissent ne pas être élevés en raison de la concentration réduite de la céruloplasmine. Cette augmentation est vraisemblablement la cause immédiate des dépôts de cuivre et de la toxicité résultante dans le cerveau et les autres tissus.2
La toxicité du cuivre dans le cerveau semble également impliquer un stress oxydatif. Des concentrations élevées de cuivre dans le milieu entourant les neurones modifient l’activité d’enzymes clés contenant le cuivre pouvant contribuer aux dommages neuronaux.1
Manifestations cliniques
La présentation clinique de la maladie est protéiforme, atteignant plusieurs organes, dont principalement le foie, le cerveau et l’œil.
Atteintes hépatiques
Elles varient largement, allant des anomalies biochimiques et la stéatose asymptomatique jusqu’à l’hépatite fulminante et l’insuffisance hépatique aiguë, en passant par l’hépatite chronique et la cirrhose.
Atteintes neurologiques
A l’image de l’atteinte hépatique, différents syndromes neurologiques sont décrits, se manifestant principalement par deux modèles : (1) un trouble du mouvement accru ou anormal, qui peut être caractérisé par des tremblements, une perte de contrôle de la motricité fine ou une dystonie, ou (2) une perte relative du mouvement qui ressemble à la rigidité parkinsonienne.1 À noter que divers troubles de la parole peuvent survenir avec l’une ou l’autre de ces manifestations.3
Atteintes psychiatriques
Les symptômes comportementaux et psychiatriques sont variables et plus fréquents chez les patients présentant une atteinte neurologique que chez ceux présentant une atteinte hépatique. La dépression est le symptôme le plus courant. Les changements de personnalité, les phobies, le comportement incongru et l’irritabilité ont été rapportés. Chez la plupart des patients présentant des caractéristiques purement psychiatriques, le diagnostic est reporté à un âge avancé (par exemple, ces manifestations peuvent être attribuées à la puberté).3.4
Atteintes oculaires
L’atteinte ophtalmique est caractérisée par l’anneau de Kayser-Fleischer : un dépôt de cuivre de couleur brune dans la membrane de Descemet de la cornée. Il est évocateur de la maladie de Wilson car observé chez environ 98% des patients présentant des manifestations neurologiques et 50% des patients présentant des manifestations hépatiques.3 Bien qu’il puisse être visible à l’œil nu, il est mieux observé à la lampe à fente. Un examen ophtalmologique doit donc être demandé en cas de suspicion de maladie de Wilson.5
Atteintes hématologiques
L’anémie hémolytique à test de Coombs négatif peut être le seul symptôme initial de la maladie de Wilson. Cependant, une hémolyse marquée est souvent associée à une maladie hépatique sévère.
Autres
La maladie de Wilson peut se manifester par des atteintes néphrologiques (syndrome de Fanconi), articulaires (arthrite) et cardiaques (cardiomégalie ; arythmies cardiaques).1
Diagnostic
Le degré de variabilité clinique de la maladie rend souvent la confirmation du diagnostic très difficile.
En règle générale, la combinaison de l’anneau de Kayser-Fleischer et d’un faible taux sérique de céruloplasmine (< 0,1 g/L) est suffisante pour établir un diagnostic. Or lorsque l’anneau de Kayser-Fleischer n’est pas présent (comme cela est courant dans la manifestation hépatique de la maladie de Wilson), le niveau de céruloplasmine n’est pas toujours fiable. Par conséquent, une combinaison de tests reflétant un métabolisme du cuivre perturbé peut être nécessaire mais aucun d’entre eux n’est per se spécifique (la céruloplasmine sérique, le cuivre sérique « libre » ou « échangeable », la cuprurie des 24 heures et le cuivre intrahépatique). Un score diagnostique basé sur tous les tests disponibles a été proposé par un groupe de travail lors de la 8e réunion internationale sur la maladie de Wilson, Leipzig 2001.10
Traitement
Administré à vie, le traitement vise principalement à corriger la surcharge en cuivre, en association avec un régime sans cuivre.
Trois armes thérapeutiques sont reconnues : la D-pénicillamine, la trientine et le zinc. La chélation avec le tétrathiomolybdate reste expérimentale.
Le traitement doit être envisagé en deux phases : (1) éliminer le cuivre tissulaire qui s’est accumulé et (2) prévenir la réaccumulation. L’élimination du cuivre est obtenue par l’administration de chélateurs puissants.
Le premier chélateur qui a été utilisé est la D-pénicillamine, cependant 30% des patients ne tolèrent pas cette thérapie à long terme. La trientine est une alternative de traitement fréquemment utilisée dans cette situation mais peut être utilisée en première intention.
La prévention de la réaccumulation peut être obtenue avec des chélateurs ou en utilisant des sels de zinc. Le zinc oral agit en empêchant l’absorption du cuivre par augmentation de la métallothionéine endogène, mais également hépatique.6
La transplantation hépatique reste l’arsenal thérapeutique souvent nécessaire pour les patients présentant une insuffisance hépatique aiguë ou une cirrhose décompensée due à la maladie de Wilson.10
Conclusion
Bien que la maladie de Wilson soit une affection rare, elle est souvent létale. Savoir l’évoquer devant des manifestations typiques peut améliorer le pronostic vital par l’utilisation d’un traitement chélateur et l’éviction alimentaire du cuivre.
Références
1- Sanyal A, Lindor K, Boyer T, Terrault N. Zakim and Boyer’s Hepatology. 7th ed. Elsevier; 2018.
2- Schilsky ML. Wilson disease: Epidemiology and pathogenesis. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com
3- Schilsky ML. Wilson disease: Clinical manifestations, diagnosis, and natural history . Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com
4- Feldman M, Friedman L, Brandt L. Sleisenger and Fordtran’s gastrointestinal and liver disease. Philadelphia, PA: Saunders/Elsevier; 2016.
5- Schilsky ML. Wilson disease: Diagnostic tests . Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com
6- Schilsky ML. Wilson disease: Treatment and prognosis. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com
7- Gu M, Cooper J, Butler P, Walker A, Mistry P, Dooley J et al. Oxidative-phosphorylation defects in liver of patients with Wilson’s disease. The Lancet. 2000;356(9228):469-474.
8- Einer C, Leitzinger C, Lichtmannegger J, Eberhagen C, Rieder T, Borchard S et al. A High-Calorie Diet Aggravates Mitochondrial Dysfunction and Triggers Severe Liver Damage in Wilson Disease Rats. Cellular and Molecular Gastroenterology and Hepatology. 2019;7(3):571-596.
9- Lang P, Schenck M, Nicolay J, Becker J, Kempe D, Lupescu A et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nature Medicine. 2007;13(2):164-170.
10- EASL Clinical Practice Guidelines: Wilson’s disease. Journal of Hepatology. 2012;56(3):671-685.