Glycogen Storage Diseases A Complex Problem Behind Complexed Sugar
Rihab FELLAH
“The ability of a cell to carry out all these interlocking metabolic processes simultaneously – obtaining every product in the amount needed and at the right time, in the face of major perturbations from outside, and without generating leftovers – is an astounding accomplishment.” Albert Lehninger, David L. Nelson, Michael M. Cox.
Glycogen metabolism, like most phenomena in the human body, has been illuminated by the different defects involving its different enzymatic steps. These enzymatic defects resulted in a group of pathologies termed glycogen storage diseases. Both pathology and physiology of glycogen metabolism were rigorously studied by the impressive couple: Carl F. Cori and Gerty T. Cori. This team of husband and wife have trained in medical studies in Europe right after World-War I (impressively, Mrs. Cori finished her premedical and medical studies in one year). Afterwards, they opened their laboratory for biochemical studies in Washington in which they pursued their research. They were subsequently awarded the Nobel prize of physiology in 1947.
In order to tackle the issue of glycogen storage diseases or GSDs, we must first explain some of the Coris lives’ work involving Glycogen synthesis and breakdown as well as their regulating mechanisms.
Glycogen metabolism
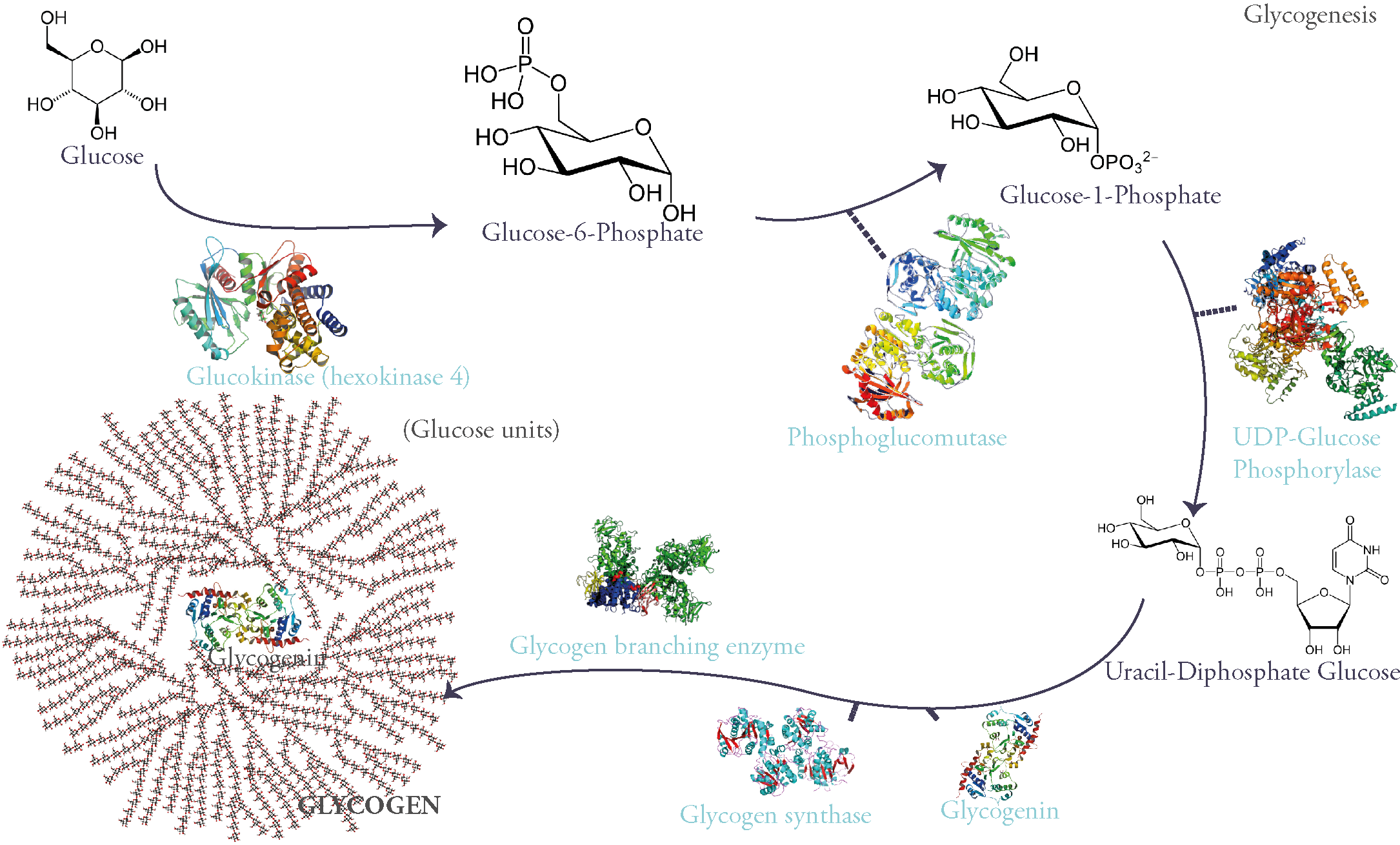
Glycogen is a branched glucose polymer that consists mainly of these monomers linked through an α 1-4 glycosidic link on the main chains. They can be secondarily branched through an α 1-6 glycosidic link. It is the most important form of glucose storage and the fastest source of energy during exercise for it provides the muscles (both skeletal and myocardial) with the necessary glucose during the first 30 minutes (i.e gluconeogenesis and other energy producing pathways take time). On the liver however falls the responsibility of maintaining stable blood glucose levels necessary for the energetic needs of all tissues during fasting. This is particularly important for certain tissues that only use glucose as fuel, as in the case of the brain.
‘Why is glycogen branched?’ you might ask. Well, because for starters it lowers the mass of water absorbed by all this sugar molecules which could disrupt the cell’s osmotic equilibrium (concentration of dissolved glucose in the cytoplasm is 0,4M while glycogen’s concentration is 0,01µM). A polymer made of a single chain of glucose could: (1) be toxic to cells, (2) limit enzymatic action on the molecule (glycogen phosphorylase and glycogen synthase can only start working from a chain’s nonreducing end). Therefore, this rose-like branched polymer of glucose gives multiple ends for enzymes to work on, is harmless for the cell and is more soluble.
Glycogen is synthesized from glucose. First glucose is phosphorylated on its 6th carbon molecule and becomes Glucose-6-Phosphate (G6P). G6P can either be used in glycolysis to produce energy or go into storage in the form of glycogen. When the latter pathway is taken G6P becomes G1P through the work of the enzyme phosphoglucomutase. G1P is then attached to a nucleotidic group called UDP (Uracyl-Di-Phosphate) therefore becoming UDP-Glucose: a sugar nucleotide, an indispensable intermediate for sugar polymerization.
These molecules are then polymerized through α 1-4 glycosidic links via the activity of the enzyme glycogen synthase.
However, this enzyme can only extend an existing glycosidic chain. That is why glycogenesis is initiated by a self-glycosylating protein called glycogenin. It is the primer of the process and links itself to 7 UDP-glucoses via an intrinsic enzyme that has a glycosyltransferase activity. Once this chain of glycogenin and 7 glucose units is formed, glycogen synthase begins adding on glucose units through α-1-4 glycosidic links.
Of course, glycogen synthase as said before can only extend starting from a non-reducing end. So, after 4 to 7 sugars in length, a Glycogen Branching Enzyme (GBE) commences the branching of a side chain from the main one by linking two glucose units by an α 1-6 glycosidic link.
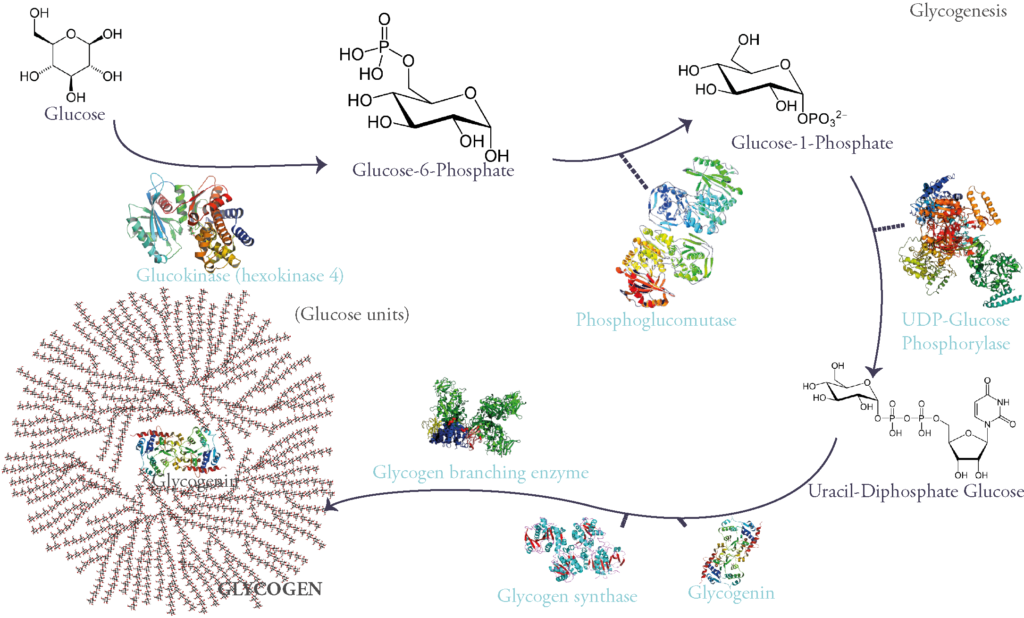
Evidently, glycogen synthesis is stimulated by insulin, the main hypoglycemic and anabolic hormone of sugar metabolism. It is therefore inhibited by glucagon and other hyperglycemic hormones such as epinephrine (aka adrenalin).
The direct opposing process of glycogenesis is glycogenolysis or glycogen breakdown. It is catalyzed by the first phosphorylase to be ever discovered (it is still referred to honorarily as THE phosphorylase in scientific literature), the famous glycogen phosphorylase (GP).
Phosphorolysis is different from hydrolysis (glycogen and starch breakdown by amylase during digestion) in the fact that, in breaking the α 1-4 glucosidic link and forming a phosphor ester such as G-1-P, it preserves some of the breakdown energy.
Phosphorylase is present in two interchangeable forms: phosphorylase a, the active catalytic form, and phosphorylase b, the quiescent form of the enzyme.
The passage from the inactive unphosphorylated form to the other active phosphorylated form is catalyzed by a phosphokinase b (PKb). This enzyme is activated by chain of reactions induced by adrenalin’s or glucagon’s binding to their membrane receptors and causing an elevation in intracellular cyclic AMP. This concept has awarded Sutherland, one of the Coris’ partners, a Nobel prize in physiology in 1971.
It is important to note that GP stops when only 4 glucose units are left to an α 1-6 branch point. Here comes the role of the Glycogen Debranching Enzyme (GDE) that can break the lateral α 1-6 bonds and provide more nonreducing ends for the phosphorylase to act on. This action requires two steps: first, GDE moves the lateral chain to the main one leaving one glucose at the branch point. This final unit is then freed alone when the α 1-6 bond is broken.
As to glycogenolysis, whether in the liver or muscle, it results in the formation of G1P and G6P through the activity of the phosphoglucomutase. Once G6P is formed, it is either glycolyzed and that occurs in the muscle; or it is transported to the endoplasmic reticulum (ER) through a specific transporter termed Glucose-6-Phosphate Transporter or T1 and then catalyzed into glucose and Pi by an enzyme specific only to the liver called glucose-6-phosphatase. Thereafter, the resulting glucose is transported to the cytoplasm and from there to the blood.
When dietary provisions are low, hepatic release of glucose after glycogen breakdown is the only way to prevent hypoglycemia, because the liver has Glucose-6-phosphatase. This enzyme is absent in adipose and muscle tissue, therefore neither can help replenish blood glucose levels.
Glycogen synthase and Glycogen phosphorylase are said to be rate-limiting enzymes of glycogenesis and glycogenolysis respectively. This signifies that they catalyze the steps where the most energy is needed for the reaction to occur and therefore this can slow the whole process by lack of ATP. They can also be controlled hormonally (via the action of hypoglycemic or hyperglycemic hormones) and allosterically (i.e when these enzymes are bound to certain molecules such as phosphate, they change conformation taking either activated or deactivated form).
Let’s explain further: glycogen phosphorylase is activated via phosphorylation by the action of glucagon and epinephrine. Glycogen synthase on the other hand, is deactivated when phosphorylated. This promotes glucose liberation and hyperglycemia. In contrast, insulin binding to its receptor activates a chain of reaction which finally induces dephosphorylation of both glycogen synthase and glycogen phophorylase, activating the former and deactivating the latter and therefore favoring glucose storage.
Another pathway through which glycogen breakdown can occur is glycophagy or autophagy. Indeed, in the lysosome exists an acid maltase that can break down α 1-4 glycosidic bonds. It is called GAA or acid α glucosidase. GAA is synthesized in ER and transported under an inactive form to the lysosome while carried by IGF2R (Insulin-like Growth Factor 2 Receptor). Its association to this specific receptor requires the existence in its composition of a certain amount of mannose 6 phosphate. Once in the lysosome, GAA is activated by the acid environment and begins breaking down polyosidic chains that have been transported into the lysosome. IGF2R is found in different levels of the endosomic-lysosmic pahways. It is also expressed in different levels on cell membranes. We’ll see later how this is important.
Why this second Glycogen breakdown pathway exists? We do not know. We are however aware that they are both necessary for the good functioning of cells and glycogen metabolism. They are complementary and any pathological defect in either can cause serious consequences as we’re about to see.
As we’re finishing with our overview of the biochemistry behind glycogen synthesis and breakdown, the readers must’ve concluded (or not, because we talked too much) that glycogen metabolism is mainly hepatic or muscular and they aren’t quite mistaken. Nonetheless, in the last two decades, scientific focus has turned towards some neurological disorders that seem to be caused by an excess of some type of glycogen in the brain. More light needs to be shed on these particularities to understand the physiologic processes governing them.
Pathology of glycogen metabolism
Glycogen storage diseases have grown in numbers throughout the years. They are numbered chronologically as of the enzymatic defect’s time of discovery. As our understanding of the underlying biochemical reactions became clearer, the classifications have changed through the years. So don’t be flustered if by chance you stumbled upon a different classification than the one we’re about to enumerate.
GSD 0 is related to the partial or total defect in glycogen synthase. GSD I, or von Gierke disease, is due to the lack of glucose-6-phosphatase or glucose-6-phosphate transporter (T1). GSD II, or Pompe’s (pronounced pompey) disease, owing to the partial or total lack of GAA. GSD III, or Cori/Forbes’ disease, resulting from the lack of the debranching enzyme. While GSD IV, or Anderson’s disease, is occasioned by the lack of the branching enzyme. GSD V and VI, or MacArdle’s and Her’s diseases, caused by a deficiency in muscular and hepatic phosphorylase respectively.
Some of the newly discovered GSDs include Lafora disease due to the pathological storage of an abnormal form of glycogen in nervous cells.
There are many more that have been added but due to issues of conciseness we unfortunately won’t be getting into them.
GSD 0: deficiency in glycogen synthase
The lack of glycogen synthase makes this pathology the only one in glycogen storage diseases where there is no glycogen storage whatsoever. Its symptoms vary depending on the mutations involved.
If the gene of muscular glycogen synthase is defective, the patient would present cardiomyopathy (disease of the cardiac muscle) and exercise intolerance. Because glycogen breakdown covers the first 30 minutes of exercise, no glucose would be released into the muscle due to the lack of glycogen and the delay it takes gluconeogenesis to produce glucose.
When hepatic glycogen synthase is absent or non-functional, the disease would have variable signs and symptoms depending on mealtimes. Postprandial time is characterized by hyperglycemia (no glucose is stored) and hyperlactatemia (erythrocytes break excess glucose without oxygen due to their lacking in mitochondria, thus producing lactate). Fasting hypoglycemia would cause cells to solicit other energy-producing pathways such as fat and protein breakdown therefore inducing hyperlipidemia, ketonic acidosis and hyperuricemia.
Diagnosis is difficult due to diversity of symptoms throughout the day but when suspected the disease would be confirmed by a muscle biopsy or DNA sequencing in leucocytes.
Treatment is mainly symptomatic by the consumption of hyperprotidic diet (to refuel gluconeogenesis), and of cornstarch at regular intervals (made of complex glucose-containing polysaccharides that are hydrolyzed slowly, permitting a gradual release of glucose that would maintain stable blood glucose levels especially during night). Patients are also advised against fasting.
GSD I: von Gierke disease
It is the lack of glucose-6-phosphotase that characterizes this entity. Since this enzyme is only present in liver and kidney, the accumulation of G6P would mainly affect these two organs causing hepatomegaly and renal failure. The inability of the liver to convert G6P to glucose is compromised, therefore it is unable to maintain stable blood glucose levels throughout the day and especially during fasting. Consequently, fasting ketonic hypoglycemia occurs and can even be complicated by hypoglycemic seizures (the brain’s favorite fuel is glucose, remember?), and just like in GSD 0, the body’s other energy producing pathways will be solicited and therefore hyperuricemia and hyperlipidemia occur.
Now, if glucose-6-phosphatase is present in the endoplasmic reticulum and no G6P can access it, the same symptoms develop. This is encountered in case of G6P transporter (T1) deficiency. Another subtype of von Gierke disease.
Diagnosis is difficult because there exists numerous similarities with other GSDs and only genetic studies can rule things out.
In this case, the treatment is mainly symptomatic too through avoiding fasting and regularly eating cornstarch. A multidisciplinary management of these patient by a hepatologist, a nephrologist and an endocrinologist is primordial.
GSD III: Cori/Forbes’ disease
This is the case, when the debranching enzyme is absent, phosphorylase would stop breaking down glycogen 4 residues away from an α 1-6 branch. Therefore, an abnormal accumulation of branched glycogen happens. This accumulation in the liver causes hepatomegaly and hepatic adenomas with growth retardation due to coma after hypoglycemia ketoacidosis and hyperlipidemia. Hepatic adenomas may favor the evolution to chronic liver disease, augmenting thus the risk of hepatocellular carcinoma.
On the other hand, accumulated branched glycogen in the muscle will cause hypertrophic cardiomyopathy that’ll lead to arrhythmia and heart failure. While skeletal muscle wasting and hypotonia as well as weakness would be present touching mainly distal musculature.
GSD IV: Anderson’s disease
It happens to affect the branching enzyme. Its defect would cause the synthesis of linear glycogen called polyglucosan. It can be developed in utero resulting in hydrops fetalis, cardiac dysfunction and consequently death. There exists infantile forms, late childhood onset forms and forms touching adults, depending on the severity of enzymatic defect.
Similarly to Cori’s disease, the accumulation of this abnormal form of glycogen causes hepatomegaly, muscular weakness and wasting; and in severe forms cardiomyopathy. Hypoglycemia isn’t an early sign of the disease but comes later in the picture as a feature of hepatic failure in its advanced forms. Neural involvement seen especially in adults due to overload of polyglucosan in both central and peripheral nervous system has been described. This form is called Adult Polyglucosan Body Disease (APBD).
The diagnosis remains genetic sequencing or histologic study of liver biopsy.
Sadly, no specific treatment exists. Symptomatic management consisting of liver transplantation has shown in some cases a slowdown in extrahepatic disease. However, in many patients, disease progresses despite transplantation. As for the neurological signs of APBD, rapamycin, a GS inhibitor, seems to diminish polyglucosan neurotoxicity in vitro, suggesting potential therapeutic effects.
GSD V and VI MacArdle’s and Her’s diseases
These entities are caused by a deficiency in muscular and hepatic phophorylase respectively.
In the former case, disease will be displayed as cramps, myalgia (mainly intermittent and exercise-induced muscular pain), fatigue and sometimes muscle swelling. Associated laboratory findings include elevation in CPK (non-specific intramuscular biochemical marker) and myoglobulinuria. Patients may describe a “second wind” phenomenon after approximately 10 minutes of exercise in which symptoms are alleviated spontaneously. That is due to energy intake from fatty acid catabolism and elevated blood flow that procures hepatic glucose. If rhabdomyolysis occurs, acute renal injury may consequently follow.
In this form of the disease, no hepatic anomaly is observed.
In the case of hepatic phosphorylase deficiency, hypoglycemia occurs associated as always with hyperlipidemia, hyperuricemia and ketonic acidosis. Growth retardation is also observed as well as hepatomegaly due to glycogen accumulation and disrupted glucose levels.
No muscular manifestation is observed though and muscular function is conserved.
Another disease may mimic either Her’s or MacArdle’s. It is the deficiency in phosphokinase b. Since glycogen phosphorylase is activated when phosphorylated by phosphokinase b, the last of a cascade of steps initiated by glucagon’s or epinephrine’s interaction with their respective membrane receptors. If phosphokinase b is deficient glycogen phosphorylase stays in its inactive form (phosphorylase b). Depending on whether the PKb affected is muscular or hepatic, disease manifestation would either resemble those observed in MacArdle’s or Her’s diseases.
Lafora disease
It is a glycogen storage disease that touches mainly the brain. It is due to the lack of Laforin, an enzyme that is thought to regulate Glycogen synthesis. When lacking, an accumulation of hyperphosphorylated glycogen in most tissues happens but particularly in nervous cells. Histologically, the build-up of this unnatural substance is characteristically observed as Lafora bodies, that ultimately damage cells.
GSD II: Pompe’s disease
Last but not least, we’re talking about Pompe’s disease. Not because we dislike it but because it is the only GSD who has specific treatment. It is also a crossroad between two groups of pathologies. Indeed, it is both a glycogen storage disease and a lysosomal storage disease.
GSD II is characterized by a defect in GAA which action requires a low pH and therefore takes place in the lysosome. This results in lysosomal accumulation of glycogen which ultimately destroys lysosomes, disrupts the lysosomal-endosomal compartment, and eventually damages the cell.
When there’s complete lack of GAA, an early onset form of the disease ensues. It involves infants and is characterized by hepatomegaly, hypertrophic cardiomyopathy and muscular weakness touching essentially proximal muscles and critical when it involves respiratory muscles. It is fatal within a few years of life if not treated.
For adults, partial GAA activity is responsible of isolated muscular manifestations of the disease without cardiac involvement.
Newborn screening for Pompe’s disease is implemented in many countries around the world because ever since 2006 enzyme replacement therapy (ERT) has revolutionized its treatment. It comes in the form of recombinant human GAA or rhGAA. This therapy helps stop disease progression in infants and adults and reduce mortality due to cardiomyopathy and respiratory muscles’ affection.
The problem remains in the fact that some patients do not respond to ERT. As we’ve mentioned above, GAA transport to lysosomes is guaranteed by its interaction with IGF2 receptor. This interaction happens because this particular receptor recognizes GAA if it is rich enough in mannose-6-phosphate. Some rhGAA either aren’t rich enough in M6P, or certain patients lack sufficient numbers of IGF2R on cell membranes to deliver it to lysosomes.
Another problem with this therapy is that it can be immunogenic and therefore be neutralized before getting to its targeting tissues.
Others speculate that sometimes lysosomal destruction is so bad that it disrupts all the endosomal-lysosomal system affecting therefore lysosomal trafficking. That damage can’t be repaired by ERT alone.
That is why gene therapy (delivering genetic information via viruses to hepatic and/or muscular cells to induce GAA synthesis) can bypass both those problems and is currently under investigation.
Chaperone therapy too seems promising. The principle of this treatment is to administer molecules that stabilize both mutant GAA and improve lysosomal trafficking in order to facilitate the delivery of recombinant GAA. Undergoing trials are investigating safety, tolerability and efficacy of these treatments.
In conclusion, glycogen storage diseases are paradoxically variable and quite similar in many aspects. They aren’t easily diagnosed because they aren’t that frequent. Additionally, no specific treatment exists to most GSDs except for Pompe’s disease. However, gene therapy seems hopefully the solution. Perhaps, one day we will be talking of a definitive therapy for all GSDs as we are doing today for Pompe’s disease. This day couldn’t come soon enough.
References
1- Craigen WJ, Darras BT. Overview of inherited disorders of glucose and glycogen metabolism. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
2- Craigen WJ .Liver phosphorylase deficiency (glycogen storage disease VI, Hers disease) .Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
3- Craigen WJ, Darras BT. Glycogen branching enzyme deficiency (glycogen storage disease IV, Andersen disease). Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
4- Craigen WJ, Darras BT. Glycogen debrancher deficiency (glycogen storage disease III). Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
5- Ellingwood S, Cheng A. Biochemical and clinical aspects of glycogen storage diseases. Journal of Endocrinology. 2018;238(3):R131-R141.
6- Kakhlon O, Glickstein H, Feinstein N, Liu Y, Baba O, Terashima T et al. Polyglucosan neurotoxicity caused by glycogen branching enzyme deficiency can be reversed by inhibition of glycogen synthase. Journal of Neurochemistry. 2013 :101-113.
7- Lehninger A, Nelson DL, Cox MM. Lehninger’s Principles of Biochemistry. Fourth edition. (2008). Chapter 15: Principles Of Metabolic Regulation: Glucose And Glycogen; pp 562-591.
8- Merritt JL II. Lysosomal acid alpha-glucosidase deficiency (Pompe disease, glycogen storage disease II, acid maltase deficiency). Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
9- Merritt JL II. Myophosphorylase deficiency (glycogen storage disease V, McArdle disease). Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
10- Nascimbeni A, Fanin M, Masiero E, Angelini C, Sandri M. Impaired autophagy contributes to muscle atrophy in glycogen storage disease type II patients. Autophagy. 2012;8(11):1697-1700.
11- Sun A. Glucose-6-phosphatase deficiency (glycogen storage disease I, von Gierke disease). Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com.
12- Vita G, Vita G, Musumeci O, Rodolico C, Messina S. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 2: diseases of motor neuron and skeletal muscle. Neurological Sciences. 2019;40(4):671-681.