21-Hydroxylase Deficiency – Overview and Algerian Perspective
Rihab FELLAH
Congenital adrenal hyperplasia (CAH) is a group of inherited defects in any step of steroidogenesis that results in a fall in cortisol levels and consequently an elevation in ACTH levels originating in adrenal hyperplasia. Deficient 21hydroxylase is the most frequent of CAHs worldwide, which has prompted the use of neonatal screening in many countries leading to early diagnosis and management. In Algeria, the main challenge faced is determining the cytogenetic characteristics of these patients in our population and estimating these diseases’ incidence and prevalence in order to establish a neonatal screening.
Introduction
CAH has been first described in 1865 by pathologist Luigi de Crecchio. He related the case of a man who, upon autopsy, was discovered to have internal female genitalia and hypertrophic adrenal glands.1,2 Today, over 150 years of research separate us from that description. Many insightful discoveries in this challenging ensemble of maladies have been made. We will review them in this article succinctly but not in an exhaustive manner.
Physiology of steroidogenesis
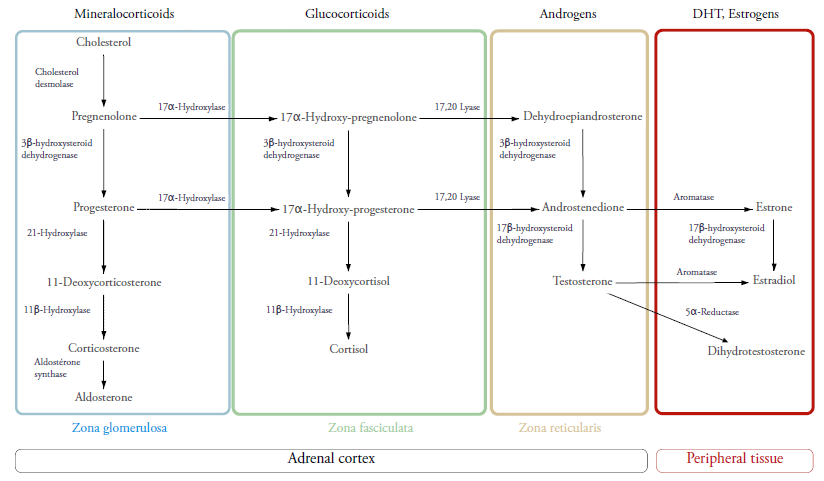
The adrenal gland is a small pyramid-like glandular tissue that rests on the posteromedial surface of the kidney. Its cortex is derived from the mesoblast. It starts functioning in the 6th week of gestation and is primarily formed of a fetal zone (FZ) and a definitive zone (DZ). The latter will give rise to the three layers or zones that are histologically and functionally different. They’re known as: zona glomerulosa (ZG), zona fasciculata (ZF) and zona reticularis (ZR). They synthesize the following major adrenocortical hormones respectively: mineralocorticoids presided by aldosterone, glucocorticoids (GC) led by cortisol and androgens particularly DHEA and its sulfated form.
Cholesterol is the original substrate of all these hormones. It is transported after specific signalling (angiotensin II, ACTH) to the mitochondria via StAR (Steroidogenic Acute Response) protein into its inner membrane where it will become pregnenolone. Depending on the zone in which the steroidogenesis is happening, specific enzymes will catalyse pregnenolone and thereafter progesterone to different steroid precursors that will lead to the main acting steroids aforementioned.1,3,4,6
In the ZG, the secretion of aldosterone is regulated by the renin-angiotensin axis stimulated by hypovolemia and hyperkaliemia. Its main function is retaining sodium and water thus causing volemic expansion. It also favours potassium release in urine to insure a sodium potassium balance.
In the ZF, cortisol, the main GC of the body (also known as the stress hormone), is regulated by the hypothalamus-pituitary-adrenal axis. Hypothalamic Corticotropin or CRH (Cortisol Releasing Hormone) is liberated during stress in the pituitary portal system. It stimulates the secretion of ACTH (Adrenal Corticotropin Hormone) by the anterior pituitary. ACTH will primarily stimulate cortisol synthesis by the ZF. Cortisol levels can also control CRH and ACTH secretion positively and negatively.
This hormone enhances cardiac output and contractility. It also elevates the heart and vasculature’s sensitivity to catecholamines. It favours glucose release through its action on lipid, glycogen and protein metabolisms therefore elevating glucose blood concentrations. To further favour this action, cortisol causes a state of insulin-resistance by stimulating catecholamine and glucagon’s excretion and by directly inhibiting glucose uptake by peripheral tissues such as muscles.
Cortisol can cause amyotrophy, incite adipocyte differentiation and adipogenesis and inhibit linear skeletal growth through its generally catabolic function. It can also give rise to depression, apathy and psychosis and is speculated to originate certain neurodegenerative diseases and memory impairment by provoking neural death in the hippocampic region.4
As for androgens, the most common among them is DHEA (DeHydroEpiAndrosterone) and DHEAS (DeHydroEpiAndrosterone Sulfate). Androgens are synthesized in the zona reticularis and converted by aromatase to estrogens, and by 5alpha-reductase to Dihydrotestosterone in peripheral tissues and minimally in the adrenal glands.5
This zona reticularis is very meagre at birth but by some unknown mechanism starts augmenting its size until it reaches its full potential by the time adrenarche occurs. This coincides with increasing levels of DHEAS throughout childhood until its peak at adrenarche permitting thus the development of axillary and pubic hair in both sexes and penile maturation in males. DHEAS concentrations then pinnacle at age 25 and eventually start declining until reverting to childhood concentrations in the last decades of life.2
The zona reticularis has the particularity of expressing cytochrome b5 in high concentrations compared to the zona fasciculata. Cytochrome b5 enhances the 17,20 lyase function of CYP17A1 that catalyses the transformation of 17OH-pregenolonone and 17OH-progesterone to DHEA and androstenedione3,4 (cf. figure 1).
The regulation of androgens’ secretion is poorly comprehended. ACTH remains the main regulatory factor of androgen synthesis. However, another factor might exist to regulate adrenarche which is yet to be discovered.4,5,7
Pathophysiology of CAH
Any defect in the enzymes catalysing any steps of steroidogenesis results in one of CAH’s diseases. These defects are all monogenic autosomal recessive mutations. They are generally heterozygotic. This here means that each of the gene’s alleles has a different mutation. The mutation that codes for the more functional enzyme is the one defining the disease.1
When an enzyme is non-functional, there’s an accumulation of precursors located upstream of the enzymatic blockade and a fall in downstream products. Some precursors can be shunted to unaffected pathways, or even take unconventional ones termed “backdoor pathways”.1,3
21 hydroxylase deficiency (21OHD)
The deficiency in 21 hydroxylase (21OHD) is the most common form of the group, accounting for 95% of CAHs.1 Indeed, the frequency of its classic form is 1 per 16000 births worldwide.2 The frequency of this enzymatic defect is important in regions where consanguine marriages are common as is the case in Turkey and the Arab Gulf countries.8 The same is speculated to be true in Algeria.9
21OH is expressed both in the ZG and ZF. It catalyses the conversion of 17OH progesterone (17OHP) to 11 deoxycortisol and Progesterone to 11 deoxycorticosterone. Its main substrate however is 17OHP, therefore its elevation is considered a good marker of enzymatic defect. That is why it is used in neonatal diagnosis through the measurement of its blood concentration via immunoassay on dried blood spots in filter paper taken upon birth.6,8
The blockade results in diminished cortisol and sometimes aldosterone activity that will stimulate in a retrograde fashion both ACTH and renin respectively. The elevation in ACTH concentration will cause adrenal hyperplasia and will further accumulate steroid precursors that will be shunted to the androgen pathway, resulting in hyperandrogenism.
Since adrenal function starts in the 6th week of gestation, this hyperandrogenism is established early in utero, coinciding with the septation of genitourinary tract into a vaginal and urethral tract in 46XX fetuses. The excess of androgens will inhibit this phenomenon. It will also act through specific androgen receptors on genital skin causing clitoral enlargement, infusion of labia folds and anterior migration of the urethral/vaginal orifice in the perineum.
The internal Wolffian (male) structures will remain absent because they require higher focal concentration of testosterone in order to develop.6,7
The classic clinical presentation of 21OHD is Disorder of Sex Development (DSD) in 46XX newborns with different degrees of sexual ambiguity as defined by the Prader scale.1,6,7,11,15,18 The different degrees of external genitalia’s virilization isn’t correlated with concentrations of androgens, but rather depends on the sensibility of their peripheral receptors which is characterized by interindividual polymorphism.6
In 46XY newborns, no DSD is present. Other signs of hyperandrogenism include precocious adrenarche, with an above average birth length and short stature in adulthood. This is due to premature epiphyseal closure, often aggravated by GC treatment.
Non-specific symptoms of mineralocorticoids’ lack vary from failure to thrive and lethargy to vomiting and poor appetite. This is the consequence of salt-wasting (SW). Biologically, the installation of hyponatremia, hyperkaliemia, hyperreninemia and even hypovolemic shock defines an adrenal crisis. It is a fatal presentation if not treated correctly.
On a psychodevelopmental level, some authors have noted that 46XX individuals with 21OHD have “male-pattern play, activities and career preferences”7 without acquiring gender-male identity if treated at birth. This is explained by the brain’s impregnation with androgens early in the fetal life.1,13
Three forms have been described depending on the degree of enzyme impairment. Classic forms manifest at birth with DSD in 46XX newborns and salt-wasting in both sexes. These forms are termed salt-wasting forms (SW-21OHD).
While other forms could manifest only with virilising disease. These patients are termed simple virilizers (SV-21OHD). The Backdoor pathway permits the conversion of 17OHP to DiHydroTestosterone (DHT) without an obligate passage through DHEA and testosterone, favouring further virilization in females with 21OHD.1,3
Last but not least, the non-classic form, whose presentation is well into adulthood, is characterized by low 17OHP levels at birth. Patients with these forms will have symptoms of latent hyperandrogenism: hirsutism and/or acne, oligomenorrhea and anovulation alongside signs of insulin resistance. Ultrasonographically, multiple cysts might be present in the ovaries making this form indistinguishable from PolyCystic Ovary Syndrome (PCOS).
In children, the Non-Classic CAH (NCCAH) appears as premature pubarche (isolated appearance of sexual hair before 8 years of age in girls and 9 years of age in boys) with or without accelerated growth velocity and advanced bone age.1,5,13
Diagnosis
21OHD is easily diagnosed when the presentation associates a DSD with salt-wasting. Treatment can be initiated without further awaiting test results especially that abstaining from treating can be dangerous.21 Nonetheless, the classic form of CAH remains underdiagnosed in boys at birth due to the lack of DSD. This explains the high frequency of adrenal crises in this class of patients and the consequent elevation of morbidity (psychomotor retardation) and mortality among them.9 This problem can be palliated with neonatal screening. It has been established in at least 17 countries. Thirteen additional countries have pilot or local screening programs.10
Diagnosis depends on measuring concentrations of 17OHP in the blood. In Algeria, the method used is radioimmunoassay found in nuclear medicine laboratories (University Hospital of Beb El Oued and Center of Pierre and Marie Curie). It is capable of reliably providing the exact concentration of this precursor. Outside of the Capital, this measurement is done in private facilities. Unfortunately, they do not meet the necessary qualification (semi-quantitative test). They therefore are not conclusive.21
If 17OHP levels are borderline, a complete adrenocortical profile after cosyntropin stimulation is recommended.11
In individuals passed their infancy, the test used is early-morning (before 8 am) baseline serum 17-hydroxyprogesterone measurement by liquid chromatography–tandem mass spectrometry.11
In case of equivocal results of the aforementioned tests, an early initiation of GC treatment or in case of genetic counseling genotyping can be performed.11
Dr Ladjouz has reported the use of genotyping in approximately 30 families affected with 21OHD.21
A cytogenetic and molecular study of DSD is being conducted by a second year doctorate student in molecular genetics at the university MENTOURI brothers in Constantine, Housna Zidoune. Its goal is to determine the implications of certain genetic and environmental factors in the development of these disorders in the Algerian population. Ms Zidoune acknowledged in a correspondence the many challenges she faces in her retrospective study. Notably, the fact that many patients lost touch with their treating physicians due to financial problems or parental denial of their children’s affliction. She recognizes the need of a more robust psychological support.20
Treatment
It consists of administering GC to reduce ACTH levels, substitute lacking cortisol and prevent shunting of excessive steroid precursors. The GC of choice is hydrocortisone (HC).1,11
Society guidelines recommend against the use of long-acting corticosteroids in adults.
In prevention of adrenal crises in newborns, fludrocortisone is the mineralocorticoid of choice, in association to sodium chloride supplements.11
In Algeria, fludrocortisone is not commercialized, though it is one of WHO’s essential medicines.22 Both parents and specialists are obliged to make ends meet in order to obtain it from neighboring countries.9,21
In NCCAH, treatment with oral contraceptives and antiandrogens are considered as first line agents. In case of failure, non-tolerance of first line agents, GC treatment is recommended.13
Parents and patients’ education is quintessential in preventing adrenal crises. Patients and guardians should learn how to recognize this emergency and prevent it through increasing GC doses11,12.
Misconceptions relating to DSD are often encountered. Parents tend to think that their children are “boys and girls” at the same time which is not the case in 46XX individuals affected with 21OHD.21 Health communication problems and lack of parent-to-parent supportive groups have been reported in several studies. Especially that caring for a child with CAH can be a financial constraint to families in the developing countries.12 This is why health associations as well as vulgarization talk-shows can help clarify the picture for them.12,21
In our country, health professionals especially pediatricians, neonatologists and mid-wives must be well-informed of this affection. This could contribute to a better management of DSD at birth and lower the mortality associated with dehydration in boys.21
Surgical treatment
In girls with virilizing disease, the timing of surgery and the exact techniques are still a matter of debate, with some specialists recommending early surgery in infancy while others advocate waiting until the child is old enough to choose for him/herself.1,11,18
Though girls with 21OHD have male-pattern behavior due to exposure to androgens in early fetal life, refusing the assigned gender rarely occurs and haven’t been proven.1,6,12 The main challenge being the complications of surgery that could cause fertility and psychosocial issues. Among which we mention, vaginal stenosis, urinary incontinence and clitoral pain.1,18
Society guidelines recommend informing the parents on the different procedures, their possible timings and the different advantages and complications of each chosen scenario.
A Jordanian study by Al-Maghribi & al (2007)14 has reported cases of females with CAH who under parental wishes, had surgical removal of internal genitalia so they would be brought up as males. Ladjouze & al (2018)9 reported that only 30,8% of their female Algerian cohort had undergone surgery, with 4 46XX patients being raised as males according to parental wishes.
We have to understand that the choice of changing the sex of one’s child is a sensible matter. Especially if the diagnosis has not been made early enough. The fear of scandal and taboo is often looming over these families. Moreover, in children with a delayed diagnosis it is more deleterious to change to the genetic sex once the sexual identity of the child is established. Therefore, maintaining a male identity in these 46XX individuals is preferable even if this dooms them fertility-wise.21
Growth-enhancing drugs
Because hyperandrogenism as well as prolonged corticoid treatment can cause adult short stature, the use of Growth Hormone (GH) alone or combined with Luteinizing Hormone-Releasing Hormone (LH-RH) agonists as well as the use of aromatase inhibitor alone or associated with antiandrogens and substitutive therapy have shown satisfying results.11,12,15
It is recommended however that these growth-enhancing drugs be used in patients whose heights are or are expected to be shorter than those of peers.11
Prenatal treatment
In mothers with high risk of conceiving children with virilizing CAH, dexamethasone administration as early as the sixth week of gestation without surpassing the eighth week can be given. This GC is chosen because it can cross the placental barrier without being metabolized. However, its safety in fetuses is controversial for it has been proven that prenatal GC can alter brain development in rodents. Adding to this dilemma is the following fact: the probability of having a female fetus affected by CAH is 1 in 8 when both parents are carrier of CAH mutation and confirming the existence of the mutation in fetal DNA found in amniotic fluid is only possible in the 20 week of gestation. This has led several medical societies to agree that prenatal treatment is experimental and should be conducted in multidisciplinary reference centers after signed consent of the parents informing them of possible adverse effects.11,16
Early non-invasive fetal DNA testing (determining fetal sex by detecting the Y chromosome in cell-free DNA present in the blood stream of the mother) promises the restriction of treatment to female fetuses.1,16 Diagnosing CAH through the same method is still under study and offers encouraging future prospects.16
In Algeria, prenatal treatment is systematically suggested to parents. It is given after an informed consent of the possible adverse effects. Ladjouze & al9 have treated over 30 mothers. SRY gene is thereafter looked for in maternal blood and if found positive (i.e. the fetus is 46XY) the treatment would be ceased. In the alternative situation, an amniocentesis is carried out in order to identify the mutation in the amniotic fluid and pursue treatment for the duration of the pregnancy. It was the case of 2 of the treated women.
When asked about the controversies regarding this treatment, Pr Ladjouze answered that given the lack of human studies proving the adverse effects of prenatal GCs on treated patients and the catastrophic social consequences that follow the birth of a child with DSD the indication of prenatal treatment is justified in our country.16,21
Improving glucocorticoids delivery methods
Single daily dose GC that minimize the number of oral takes per day while mimicking the circadian rhythm of cortisol is being tested.1,11 Once-daily modified-release oral HC in multiparticulate capsules is going through phase 3 randomized trial.11
New prospects
Because these diseases are monogenic,1 treatment with stem cells and gene editing methods is tempting and offers a definitive solution to the problem at hand.1,17
Conclusion
Diving into this intricate web of hormonal anomalies, confusion is unavoidable. One can only wonder how being affected with such unfathomable disease must feel like. The stigma this disease carries is also of great magnitude. With over 85% of affected patients and caregivers believing that classifying CAH as a DSD badly impacts CAH community,19 some authors18 suggest categorizing CAH under Adrenal insufficiency or as a standalone group altogether. In either case shedding light and not shadow on CAH in communities worldwide as well as our own is our responsibility as future health professionals and primary care givers.
References
1- El-Maouche D, Arlt W, Merke D. Congenital adrenal hyperplasia. The Lancet. 2017;390(10108):2194-2210.
2- Turcu A, Auchus R. The next 150 years of congenital adrenal hyperplasia. The Journal of Steroid Biochemistry and Molecular Biology. 2015;153:63-71.
3- Miller W. MECHANISMS IN ENDOCRINOLOGY: Rare defects in adrenal steroidogenesis. European Journal of Endocrinology. 2018;179(3):R125-R141.
4- Stewart PM, Newell-Price JDC. The Adrenal Cortex. In: Williams textbook of endocrinology edited by Melmed S, Polonsky KS, Larsen PR, Kronenberg HM—Thirteenth edition: pp 490-555.
5- Styne DM, Grumbach MM. Physiology and Disorders of Puberty. In: Williams textbook of endocrinology edited by Melmed S, Polonsky KS, Larsen PR, Kronenberg HM—Thirteenth edition: pp 1047-1218.
6- White P, Speiser P. Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency1. Endocrine Reviews. 2000;21(3):245-291.
7- Turcu A, Auchus R. Adrenal Steroidogenesis and Congenital Adrenal Hyperplasia. Endocrinology and Metabolism Clinics of North America. 2015;44(2):275-296.
8- Güran T, Tezel B, Gürbüz F, Selver Eklioðlu B, Hatipoðlu N, Kara C et al. Neonatal Screening for Congenital Adrenal Hyperplasia in Turkey: A Pilot Study with 38,935 Infants. Journal of Clinical Research in Pediatric Endocrinology. 2019;11(1):13-23.
9- Ladjouze A, Yala I, Yahiaoui M, Zerguini D et al. Age At Diagnosis And Outcome In Maghreb Patients With 21-Hydroxylase Deficient Congenital Adrenal Hyperplasia; Urgent Need For Newborn Screening. Poster presented at: 57th ESPE ; 27-29 sept 2018 ; Athens, Greece.
10- White P. Neonatal screening for congenital adrenal hyperplasia. Nature Reviews Endocrinology. 2009;5(9):490-498. 11- Speiser P, Arlt W, Auchus R, Baskin L, Conway G, Merke D et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society* Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2018;103(11):4043-4088.
12- Fleming L, Van Riper M, Knafl K. Management of Childhood Congenital Adrenal Hyperplasia—An Integrative Review of the Literature. Journal of Pediatric Health Care. 2017;31(5):560-577.
13- Martin K, Anderson R, Chang R, Ehrmann D, Lobo R, Murad M et al. Evaluation and Treatment of Hirsutism in Premenopausal Women: An Endocrine Society* Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2018;103(4):1233-1257.
14- Al-Maghribi, H. Congenital adrenal hyperplasia: Problems with developmental anomalies of the external genitalia and sex assignment. Saudi Journal of Kidney Diseases and Trans- plantation.2007; 18: 405-413.
15- Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11-ßhydroxylase deficiency. Endocrine. 2016;55(1):19-36.
16- Bachelot A, Grouthier V, Courtillot C, Dulon J, Touraine P. MANAGEMENT OF ENDOCRINE DISEASE: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: update on the management of adult patients and prenatal treatment. European Journal of Endocrinology. 2017;176(4):R167-R181.
17- Lin-Su K, Lekarev O, Poppas D, Vogiatzi M. Congenital adrenal hyperplasia patient perception of ‘disorders of sex development’ nomenclature. International Journal of Pediatric Endocrinology. 2015;2015(1).
18- Wang L, Poppas D. Surgical outcomes and complications of reconstructive surgery in the female congenital adrenal hyperplasia patient: What every endocrinologist should know. The Journal of Steroid Biochemistry and Molecular Biology. 2017;165:137-144.
19- Ruiz-Babot G, Hadjidemetriou I, King P, Guasti L. New Directions for the Treatment of Adrenal Insufficiency. Frontiers in Endocrinology. 2015;6.
20- Zidoune H. Email sent to: Rihab Fellah. 25th-26th March 2019.
21- Ladjouze A.Email sent to Rihab Fellah. 31st March 2019.
22- WHO Model Lists of Essential Medicines [Internet]. World Health Organization. 2011 [cited 3 April 2019]. Available from: http://www.who.int/medicines/publications/essentialmedicines/en/ index.html.